Approximate time: 45 minutes
Learning objectives
- Differentiate between query-sorted and coordinate-sorted alignment files
- Describe and remove duplicate reads
- Process a raw SAM file for input into a BAM for GATK
Alignment file processing
The alignment files that come from bwa are raw alignment and need some processing before they can be used for variant calling. While all of the data is present, we need to format it in a way that helps the variant calling algorithm process it. This process is very similar to flipping all of the pieces of a puzzle over to the correct side and grouping edge pieces or pieces by a similar color or pattern when starting a jigsaw puzzle.
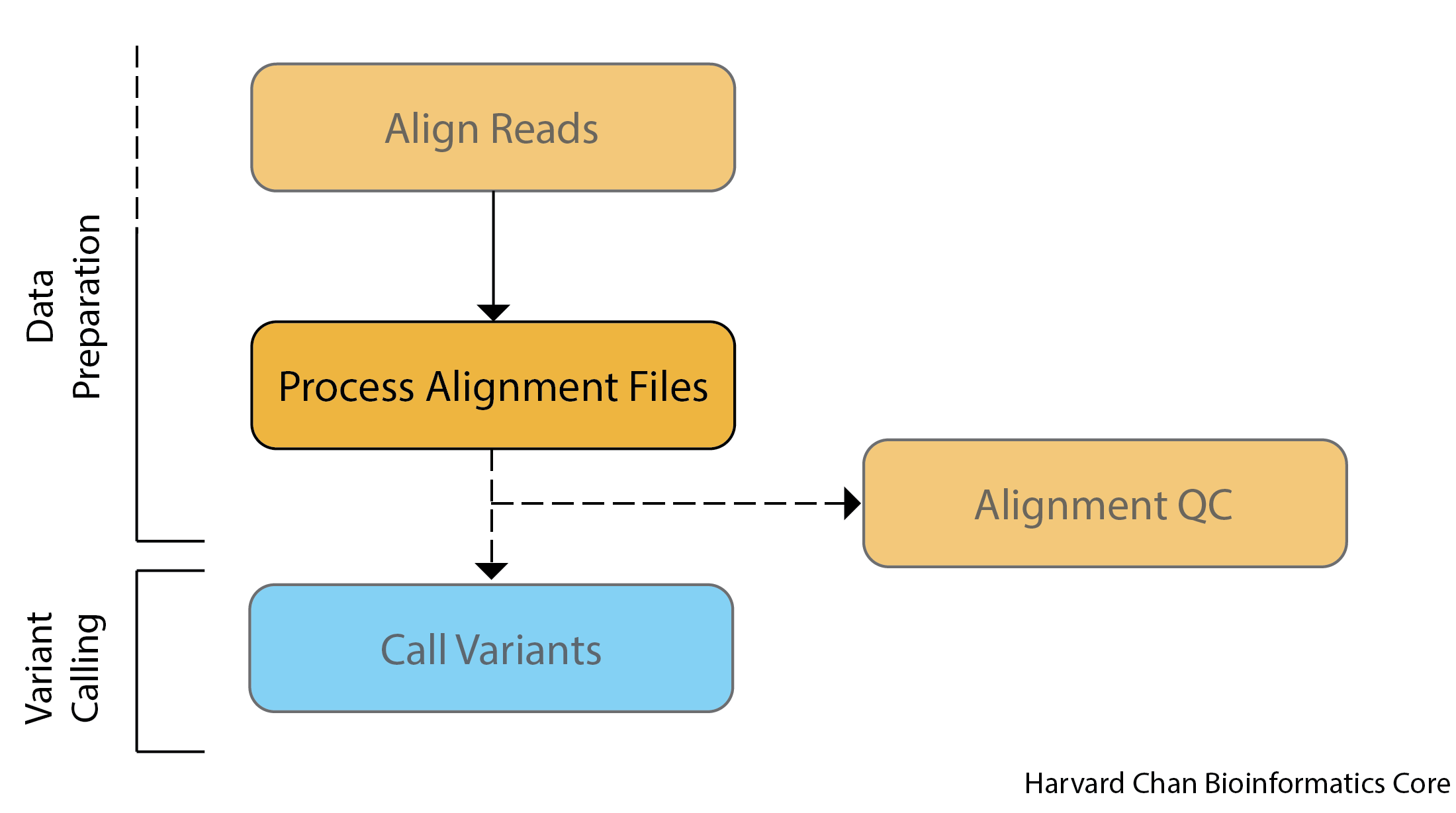
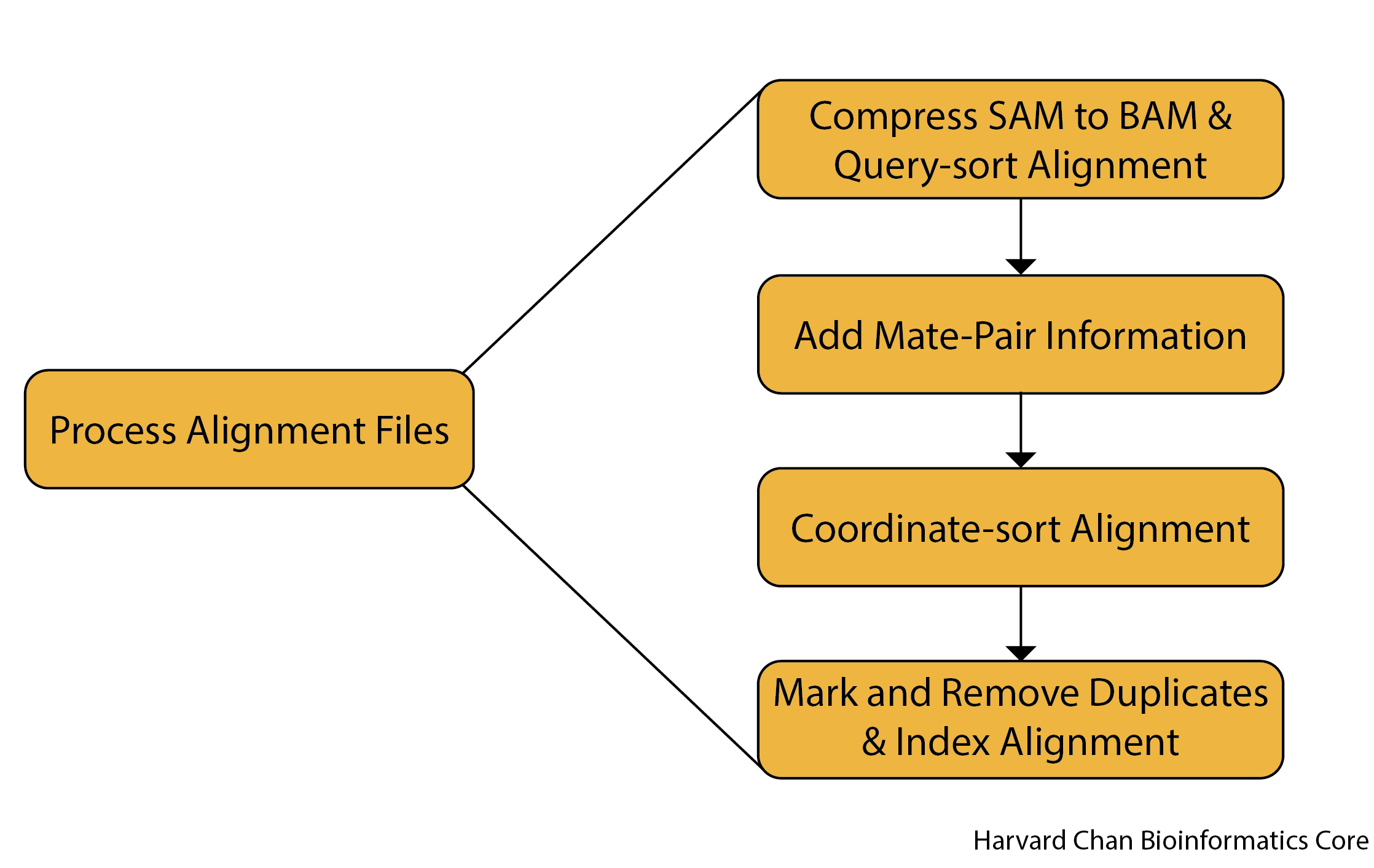
Pipeline for processing alignment files with Picard
Picard is a set of command line tools for processing high-throughput sequencing (HTS) data and formats such as SAM/BAM/CRAM and VCF. It is maintained by the Broad Institute, and is open-source under the MIT license and free for all uses. Picard is written in Java and does not have functionality for multi-threading.
Why not use
samtools?The processing of the alignment files (SAM/BAM files) can also be done with
samtools. While there are some advantages to using samtools (i.e. more user-friendly, multi-threading capability), there are slight formatting differences which we may want to take advantage of downstream. Since we will be using GATK later in this workshop (also from the Broad Institute), Picard seemed like a more suitable fit.Near the end of the lesson, there will be a dropdown, if you would like to know how to do the processing steps in
samtools.
NOTE: You may encounter a situation where your reads from a single sample were sequenced across different lanes/machines. As a result, each alignment will have different read group IDs, but the same sample ID (the SM tag in your SAM/BAM file). You will need to merge these files here before continuing. The dropdown menu below will detail how to do this.
Click here if you need to merge alignment files from the same sample
You can merge alignment files with different read group IDs from the same sample in bothPicardandsamtools. In the dropdowns below we will outline each method:Click to see how to merge SAM/BAM files in
First, we need to load thePicardPicardmodule:module load picard/2.27.5We can define our variables as:INPUT_BAM_1=Read_group_1.bam INPUT_BAM_2=Read_group_2.bam MERGED_OUTPUT=Merged_output.bamHere is the command we would need to run to merge the SAM/BAM files:java -jar $PICARD/picard.jar MergeSamFiles \ --INPUT $INPUT_BAM_1 \ --INPUT $INPUT_BAM_2 \ --OUTPUT $MERGED_OUTPUTWe can breakdown this command:
java -jar $PICARD/picard.jar MergeSamFilesThis calls theMergeSamFilesfrom withinPicard--INPUT $INPUT_BAM_1This is the first SAM/BAM file that we would like to merge.--INPUT $INPUT_BAM_2This is the second SAM/BAM file that we would like to merge. We can continue to add--INPUTlines as needed.--OUTPUT $MERGED_OUTPUTThis is the output merged SAM/BAM file
Click to see how to merge SAM/BAM files in
First, we need to load thesamtoolssamtoolsmodule, which also requiresgccto be loaded:module load gcc/6.2.0 module load samtools/1.15.1We can define our variables as:INPUT_BAM_1=Read_group_1.bam INPUT_BAM_2=Read_group_2.bam MERGED_OUTPUT=Merged_output.bam THREADS=8Here is the command we would need to run to merge the SAM/BAM files:samtools merge \ -o $MERGED_OUTPUT \ $INPUT_BAM_1 \ $INPUT_BAM_2 \ --output-fmt BAM \ -@ $THREADSWe can break down this command:
samtools mergeThis calls themergepackage withinsamtools.-o $MERGED_OUTPUTThis is the merged output file.$INPUT_BAM_1This is the first SAM/BAM file that we would like to merge.$INPUT_BAM_2This is the second SAM/BAM file that we would like to merge. We can continue to add additional input SAM/BAM files to this list as needed.--output-fmt BAMThis specifies the output format asBAM. If for some reason you wanted aSAMoutput file then you would use--output-fmt SAMinstead.-@ $THREADSThis specifies the number of threads we want to use for this process. We are using 8 threads in this example, but this could be different depending on the parameters that you would like to use.
Before we start processing our alignment SAM file with Picard, let’s take a quick look at the steps involved in this pipeline.
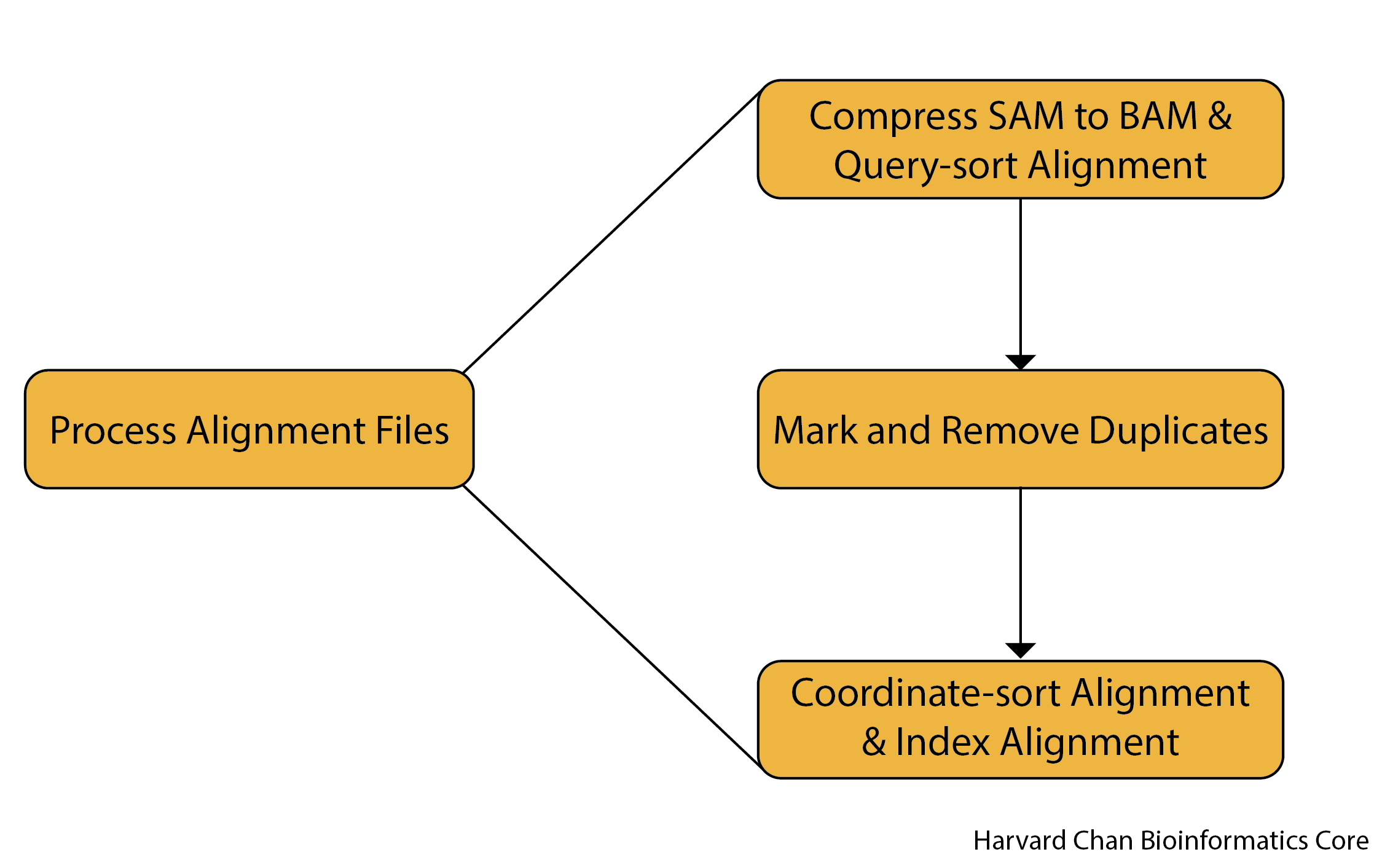
Several goals need to be accomplished, and we will go through each in detail in this lesson!
Let’s begin by creating a script for alignment processing. Make a new sbatch script within vim:
$ cd ~/variant_calling/scripts/
$ vim picard_alignment_processing_normal.sbatch
As always, we start the sbatch script with our shebang line, description of the script and our sbatch directives to request appropriate resources from the O2 cluster.
#!/bin/bash
# This sbatch script is for processing the alignment output from bwa and preparing it for use in GATK using Picard
# Assign sbatch directives
#SBATCH -p priority
#SBATCH -t 0-04:00:00
#SBATCH -c 1
#SBATCH --mem 32G
#SBATCH -o picard_alignment_processing_normal_%j.out
#SBATCH -e picard_alignment_processing_normal_%j.err
Next we load the Picard module:
# Load module
module load picard/2.27.5
Note: Picard is software that does NOT require gcc/6.2.0 to also be loaded
Next, let’s define some variables that we will be using:
# Assign file paths to variables
SAMPLE_NAME=syn3_normal
SAM_FILE=/n/scratch/users/${USER:0:1}/${USER}/variant_calling/alignments/${SAMPLE_NAME}_GRCh38.p7.sam
REPORTS_DIRECTORY=/home/${USER}/variant_calling/reports/picard/${SAMPLE_NAME}/
QUERY_SORTED_BAM_FILE=`echo ${SAM_FILE%sam}query_sorted.bam`
REMOVE_DUPLICATES_BAM_FILE=`echo ${SAM_FILE%sam}remove_duplicates.bam`
METRICS_FILE=${REPORTS_DIRECTORY}/${SAMPLE_NAME}.remove_duplicates_metrics.txt
COORDINATE_SORTED_BAM_FILE=`echo ${SAM_FILE%sam}coordinate_sorted.bam`
Finally, we can also make a directory to hold the Picard reports:
# Make reports directory
mkdir -p $REPORTS_DIRECTORY
1. Compress SAM file to BAM file
As you might suspect, because SAM files hold alignment information for all of the reads in a sequencing run and there are oftentimes millions of sequence reads, SAM files are very large and cumbersome to store. As a result, SAM files are often stored in a binary compressed version called a BAM file. Most software packages are agnostic to this difference and will accept both SAM and BAM files, despite BAM files not being human readable. It is generally considered best practice to keep your data in BAM format if it is being stored for long periods of time, unless you specifically need the SAM version of the alignment, in order to reduce unnecessary storage on a shared computing cluster.
This step will happen in the same line code used for query-sorting (below).
2. Query-sort alignment file
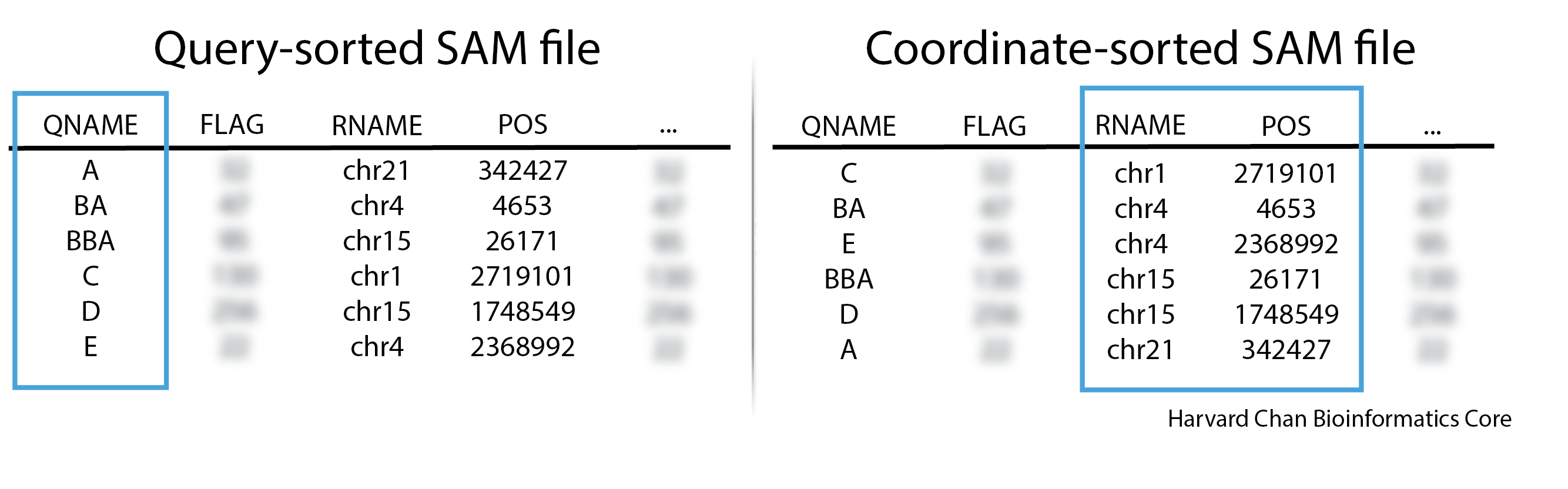
Alignment files are initally ordered by the order of the reads in the FASTQ file, which is not particularly useful. Picard can more exhaustively look for duplicates if the file is sorted by read-name (query-sorted). Oftentimes, when people discuss sorted BAM/SAM files, they are refering to coordinate-sorted BAM/SAM files.
- Query-sorted BAM/SAM files are sorted based upon their read names and ordered lexiographically
- Coordinate-sorted BAM/SAM files are sorted by their aligned sequence name (chromosome/linkage group/scaffold) and position
Picard can mark and remove duplicates in either coordinate-sorted or query-sorted BAM/SAM files, however, if the alignments are query-sorted it can test secondary alignments for duplicates. A brief discussion of this nuance is discussed in the MarkDuplicates manual of Picard. As a result, we will first query-sort our SAM file and convert it to a BAM file.
While we query-sort the reads, we are also going to convert our SAM file to a BAM file. We don’t need to specify this conversion explicitly, because Picard will make this change by interpreting the file extensions that we provide in the INPUT and OUTPUT file options.
Add the following command to our script:
# Query-sort alginment file and convert to BAM
java -jar $PICARD/picard.jar SortSam \
--INPUT $SAM_FILE \
--OUTPUT $QUERY_SORTED_BAM_FILE \
--SORT_ORDER queryname
The components of this command are:
java -jar $PICARD/picard.jar SortSamCalls Picard’sSortSamsoftware package--INPUT $SAM_FILEThis is where we provide the SAM input file--OUTPUT $QUERY_SORTED_BAM_FILEThis is the BAM output file.--SORT_ORDER querynameThe options here are eitherquerynameorcoordinate.
Why does this command look different from the Picard documentation?
The syntax that Picard uses is quite particular and the syntax shown in the documentation is not always consistent. There are two main ways for providing input for Picard: Traditional and New (Barcalay) Syntax. Commands written in either syntax are equally valid and produce the same output. To better understand the different syntax, we recommend you take a look at this short lesson.
3. Mark and Remove Duplicates
An important step in processing a BAM file is to mark and remove PCR duplicates. These PCR duplicates can introduce artifacts because regions that have preferential PCR amplification could be over-represented. These reads are flagged by having identical mapping locations in the BAM file. Importantly, it is impossible to distinguish between PCR duplicates and identical fragments. However, one can reduce the latter by doing paired-end sequencing and providing appropriate amounts of input material.
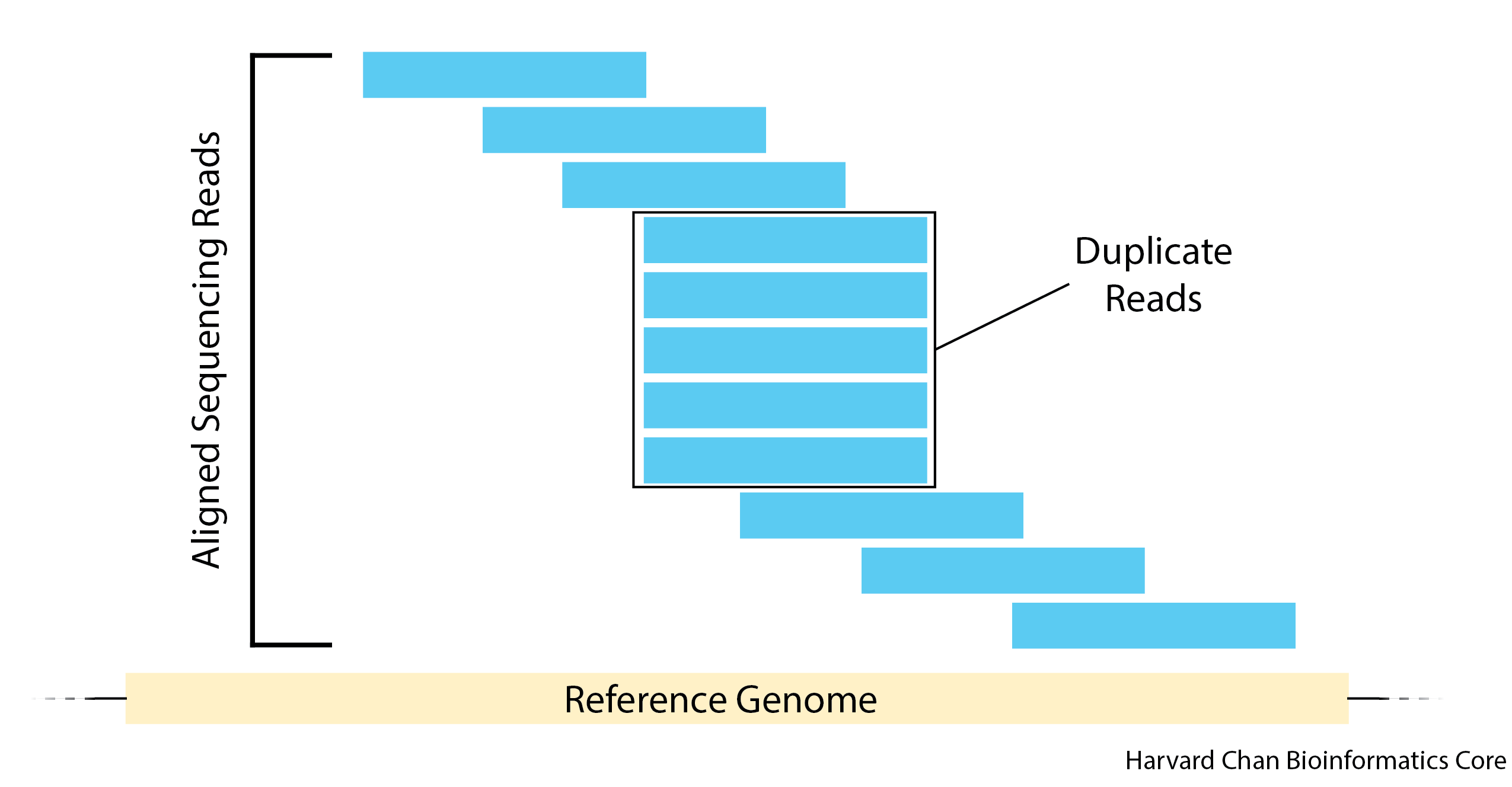
Now we will add the command to our script that allows us to mark and remove duplicates in Picard:
# Mark and remove duplicates
java -jar $PICARD/picard.jar MarkDuplicates \
--INPUT $QUERY_SORTED_BAM_FILE \
--OUTPUT $REMOVE_DUPLICATES_BAM_FILE \
--METRICS_FILE $METRICS_FILE \
--REMOVE_DUPLICATES true
The components of this command are:
java -jar $PICARD/picard.jar MarkDuplicatesCallsPicard’sMarkDuplicatesprogram--INPUT $QUERY_SORTED_BAM_FILEUses our query-sorted BAM file as input--OUTPUT $REMOVE_DUPLICATES_BAM_FILEWrites the output to a BAM file--METRICS_FILE $METRICS_FILECreates a metrics file (required byPicard MarkDuplicates)--REMOVE_DUPLICATES trueNot only are we going to mark/flag our duplicates, we can also remove them. By setting theREMOVE_DUPLICATESparameter equal totrue, we can remove the duplicates.
4. Coordinate-sort the Alignment File
For most downstream processes, coordinate-sorted alignment files are required. As a result, we will need to change our alignment file from being query-sorted to being coordinate-sorted and we will once again use the SortSam command within Picard to accomplish this. Since this BAM file will be the final BAM file that we make and will use for downstream analyses, we will need to create an index for it at the same time. The command we will be using for coordinate-sorting and indexing our BAM file is:
# Coordinate-sort BAM file and create BAM index file
java -jar $PICARD/picard.jar SortSam \
--INPUT $REMOVE_DUPLICATES_BAM_FILE \
--OUTPUT $COORDINATE_SORTED_BAM_FILE \
--SORT_ORDER coordinate \
--CREATE_INDEX true
The components of this command are:
java -jar $PICARD/picard.jar SortSamCallsPicard’sSortSamprogram--INPUT $REMOVE_DUPLICATES_BAM_FILEOur BAM file once we have removed the duplicate reads. NOTE: Even though the software is calledSortSam, it can use BAM or SAM files as input and also BAM or SAM files as output.--OUTPUT $COORDINATE_SORTED_BAM_FILEOur BAM output file sorted by coordinates.--SORT_ORDER coordinateSort the output file by coordinates--CREATE_INDEX trueSetting theCREATE_INDEXequal totruewill create an index of the final BAM output. The index creation can also be accomplished by using theBuildBamIndexcommand withinPicard, but thisCREATE_INDEXfunctionality is built into manyPicardfunctions, so you can often use it at the last stage of processing your BAM file to save having to runBuildBamIndexafter.
Go ahead and save and quit. Don’t run it just yet!
Click here to see what our final sbatchcode script for the normal sample should look like
#!/bin/bash # This sbatch script is for processing the alignment output from bwa and preparing it for use in GATK using Picard
# Assign sbatch directives #SBATCH -p priority #SBATCH -t 0-04:00:00 #SBATCH -c 1 #SBATCH --mem 32G #SBATCH -o picard_alignment_processing_normal_%j.out #SBATCH -e picard_alignment_processing_normal_%j.err
# Load module module load picard/2.27.5
# Assign file paths to variables SAMPLE_NAME=syn3_normal SAM_FILE=/n/scratch/users/${USER:0:1}/${USER}/variant_calling/alignments/${SAMPLE_NAME}_GRCh38.p7.sam REPORTS_DIRECTORY=/home/${USER}/variant_calling/reports/picard/${SAMPLE_NAME}/ QUERY_SORTED_BAM_FILE=`echo ${SAM_FILE%sam}query_sorted.bam` REMOVE_DUPLICATES_BAM_FILE=`echo ${SAM_FILE%sam}remove_duplicates.bam` METRICS_FILE=${REPORTS_DIRECTORY}/${SAMPLE_NAME}.remove_duplicates_metrics.txt COORDINATE_SORTED_BAM_FILE=`echo ${SAM_FILE%sam}coordinate_sorted.bam`
# Make reports directory mkdir -p $REPORTS_DIRECTORY
# Query-sort alginment file and convert to BAM java -jar $PICARD/picard.jar SortSam \ --INPUT $SAM_FILE \ --OUTPUT $QUERY_SORTED_BAM_FILE \ --SORT_ORDER queryname
# Mark and remove duplicates java -jar $PICARD/picard.jar MarkDuplicates \ --INPUT $QUERY_SORTED_BAM_FILE \ --OUTPUT $REMOVE_DUPLICATES_BAM_FILE \ --METRICS_FILE $METRICS_FILE \ --REMOVE_DUPLICATES true
# Coordinate-sort BAM file and create BAM index file java -jar $PICARD/picard.jar SortSam \ --INPUT $REMOVE_DUPLICATES_BAM_FILE \ --OUTPUT $COORDINATE_SORTED_BAM_FILE \ --SORT_ORDER coordinate \ --CREATE_INDEX true
Do I need to add read groups?
Some pipelines will have you add read groups while procressing your alignment files. It is usually not necessary because you can typically do it during alignment. If you are needing to add read groups, we recommend doing it first (before all the processing steps outlined above). You can use Picard
AddOrReplaceReadGroups, which has the added benefit of allowing you to also sort your alignment file (our first step anyways) in the same step as adding the read group information. The dropdown below discusses how to add or replace read groups withinPicard.Click here if you need to add or replace read groups using
In order to add or replace read groups, we are going to usePicardPicard'sAddOrReplaceReadGroupstool. First we would need to load thePicardmodule:# Load module module load picard/2.27.5The general syntax forAddOrReplaceReadGroupsis:# Add or replace read group information java -jar $PICARD/picard.jar AddOrReplaceReadGroups \ --INPUT $SAM_FILE \ --OUTPUT $BAM_FILE \ --RGID $READ_GROUP_ID \ --RGLB $READ_GROUP_LIBRARY \ --RGPL $READ_GROUP_PLATFORM \ --RGPU $READ_GROUP_PLATFORM_UNIT \ --RGSM $READ_GROUP_SAMPLEWe discussed the Read Group tags previously in the Sequence Alignment Theory and more information on them can be found here. If you would also sort your SAM/BAM file at the same time, you just need to add the
java -jar $PICARD/picard.jar AddOrReplaceReadGroupsThis calls theAddOrReplaceReadGroupspackage withinPicard--INPUT $SAM_FILEThis is your input file. It could be a BAM/SAM alignment file, but because we recommend doing this first if you need to do it, this would be a SAM file. You don't need to specifiy that it is a BAM/SAM file,Picardwith figure that out from the provided extension.--OUTPUT $BAM_FILEThis would be your output file. It could be BAM/SAM, but you would mostly likely pick BAM because you'd like to save space on the cluster. You don't need to specifiy that it is a BAM/SAM file,Picardwith figure that out from the provided extension.--RGID $READ_GROUP_IDThis is your read group ID and must be unique--RGLB $READ_GROUP_LIBRARYThis is your read group library--RGPL $READ_GROUP_PLATFORMThis is the platform used for the sequencing--RGPU $READ_GROUP_PLATFORM_UNITThis is the unit used to do the sequencing--RGSM $READ_GROUP_SAMPLEThis is the sample name that the sequencing was done on--SORT_ORDERoption to your command. If you don't add it, it will leave your reads in the same order as they were provided. The main two sort orders to be aware of are query-sorted and coordinate-sorted. A full discussion of them can be found above. If you wanted the output to be query-sorted, then you could use:--SORT_ORDER querynameOr if you wanted them to be coordinate-sorted than you could use:--SORT_ORDER coordinate
Click here for alignment file processing using Samtools
Samtools is another popular tool used for processing BAM/SAM files. The output from Samtools compared to Picard is largely the same. Below is the pipeline and explanation for how you would carry out the similar SAM/BAM processing steps within Samtools.
Click here for setting up a
sbatchscript BAM/SAM Processing for theSamtoolspipelineSetting up
First, we are going to navigate to our scirpts folder and open a newsbatchScriptsbatchsubmission script invim:cd ~/variant_calling/scripts/ vim samtools_processing_normal.sbatch
Next, we are going to need to set-up oursbatchsubmission script with our shebang line, description,sbatchdirectives, modules to load and file variables.#!/bin/bash # This sbatch script is for processing the alignment output from bwa and preparing it for use in GATK using Samtools
# Assign sbatch directives #SBATCH -p priority #SBATCH -t 0-04:00:00 #SBATCH -c 8 #SBATCH --mem 16G #SBATCH -o samtools_processing_normal_%j.out #SBATCH -e samtools_processing_normal_%j.err
# Load modules module load gcc/6.2.0 module load samtools/1.15.1
# Assign file paths to variables SAM_FILE=/n/scratch/users/${USER:0:1}/${USER}/variant_calling/alignments/syn3_normal_GRCh38.p7.sam QUERY_SORTED_BAM_FILE=`echo ${SAM_FILE%sam}query_sorted.bam` FIXMATE_BAM_FILE=`echo ${QUERY_SORTED_BAM_FILE%query_sorted.bam}fixmates.bam` COORDINATE_SORTED_BAM_FILE=`echo ${QUERY_SORTED_BAM_FILE%query_sorted.bam}coordinate_sorted.bam` REMOVED_DUPLICATES_BAM_FILE=`echo ${QUERY_SORTED_BAM_FILE%query_sorted.bam}removed_duplicates.bam`
Click here for query-sorting a SAM file and converting it to BAM for the
Similarly toSamtoolspipelinePicard, we are going to need to initally query-sort our alignment. We are also going to be converting the SAM file into a BAM file at this step. Also similarly toPicard, we don't need to specify that our input files are BAM or SAM files.Samtoolswill use the extensions you provide it in your file names as guidance for whether you are providing it a BAM/SAM file. Below is the code we will use to query-sort our SAM file and convert it into a BAM file:
# Sort SAM file and convert it to a query name sorted BAM file samtools sort \ -@ 8 \ -n \ -o $QUERY_SORTED_BAM_FILE \ $SAM_FILE
The components of this line of code are:samtools sortThis calls the sort function withinsamtools.-@ 8This tellssamtoolsto use 8 threads when it multithreads this task. Since we requested 8 cores for thissbatchsubmission, let's go ahead and use them all.-nThis argument tellssamtools sortto sort by read name as opposed to the default sorting which is done by coordinate.-O bamThis is declaring the output format of.bam.-o $QUERY_SORTED_BAM_FILEThis is abashvariable that holds the path to the output file of thesamtools sortcommand.$SAM_FILEThis is abashvariable holding the path to the input SAM file.
Click here for fixing mate information for the
Next, we are going to add more mate-pair information to the alignments including the insert size and mate pair coordinates. It is important to note that for this commandSamtoolspipelinesamtoolsrelies on positional parameters for assigning the input and output BAM files. In this case the input BAM file ($QUERY_SORTED_BAM_FILE) needs to come before the output file ($FIXMATE_BAM_FILE):# Score mates samtools fixmate \ -@ 8 \ -m \ $QUERY_SORTED_BAM_FILE \ $FIXMATE_BAM_FILE
The parts of this command are:samtools fixmateThis calls thefixmatecommand insamtools-@ 8This tellssamtoolsto use 8 threads when it multithreads this task.-mThis will add the mate score tag that will be critically important later forsamtools markdup$QUERY_SORTED_BAM_FILEThis is our input BAM file$FIXMATE_BAM_FILEThis is our output BAM file
Click here for coordinate-sorting a BAM file for the
Now that we have added theSamtoolspipelinefixmateinformation, we need to coordinate-sort the BAM file. We can do that by:# Sort BAM file by coordinate samtools sort \ -@ 8 \ -o $COORDINATE_SORTED_BAM_FILE \ $FIXMATE_BAM_FILE
We have gone through all of the these paramters already in the previoussamtools sortcommand. The only difference in this command is that we are not using the-noption, which tellssamtoolsto sort by read name. Now, we are excluding this and thus sorting by coordinates, the default setting.
Click here for marking and removing duplicates for the
Now we are going to mark and remove the duplicate reads:Samtoolspipeline# Mark and remove duplicates and then index the output file samtools markdup \ -r \ --write-index \ -@ 8 \ $COORDINATE_SORTED_BAM_FILE \ ${REMOVED_DUPLICATES_BAM_FILE}##idx##${REMOVED_DUPLICATES_BAM_FILE}.baiThe components of this command are:samtools markdupThis calls the mark duplicates software insamtools-rThis removes the duplicate reads--write-indexThis writes an index file of the output BAM file-@ 8This sets that we will be using 8 threads$BAM_FILEThis is our input BAM file${REMOVED_DUPLICATES_BAM_FILE}##idx##${REMOVED_DUPLICATES_BAM_FILE}.baiThis has two parts:- The first part (
${REMOVED_DUPLICATES_BAM_FILE}) is our BAM output file with the duplicates removed from it - The second part (
##idx##${REMOVED_DUPLICATES_BAM_FILE}.bai) is a shortcut to creating a.baiindex of the BAM file. If we use the--write-indexoption without this second part, it will create a.csiindex file..baiindex files are a specific type of.csifiles, so we need to specify it with the second part of this command to ensure that a.baiindex file is created rather than a.csiindex file.
- The first part (
Click here for the final normal sample
The final script should look like:sbatchscript to do the BAM/SAM processing for theSamtoolspipeline#!/bin/bash # This sbatch script is for processing the alignment output from bwa and preparing it for use in GATK using Samtools
# Assign sbatch directives #SBATCH -p priority #SBATCH -t 0-04:00:00 #SBATCH -c 8 #SBATCH --mem 16G #SBATCH -o samtools_processing_normal_%j.out #SBATCH -e samtools_processing_normal_%j.err
# Load modules module load gcc/6.2.0 module load samtools/1.15.1
# Assign file paths to variables SAM_FILE=/n/scratch/users/${USER:0:1}/${USER}/variant_calling/alignments/syn3_normal_GRCh38.p7.sam QUERY_SORTED_BAM_FILE=`echo ${SAM_FILE%sam}query_sorted.bam` FIXMATE_BAM_FILE=`echo ${QUERY_SORTED_BAM_FILE%query_sorted.bam}fixmates.bam` COORDINATE_SORTED_BAM_FILE=`echo ${QUERY_SORTED_BAM_FILE%query_sorted.bam}coordinate_sorted.bam` REMOVED_DUPLICATES_BAM_FILE=`echo ${QUERY_SORTED_BAM_FILE%query_sorted.bam}removed_duplicates.bam`
# Sort SAM file and convert it to a query name sorted BAM file samtools sort \ -@ 8 \ -n \ -o $QUERY_SORTED_BAM_FILE \ $SAM_FILE
# Score mates samtools fixmate \ -@ 8 \ -m \ $QUERY_SORTED_BAM_FILE \ $FIXMATE_BAM_FILE
# Sort BAM file by coordinate samtools sort \ -@ 8 \ -o $COORDINATE_SORTED_BAM_FILE \ $FIXMATE_BAM_FILE
# Mark and remove duplicates and then index the output file samtools markdup \ -r \ --write-index \ -@ 8 \ $COORDINATE_SORTED_BAM_FILE \ ${REMOVED_DUPLICATES_BAM_FILE}##idx##${REMOVED_DUPLICATES_BAM_FILE}.bai
Click here for the final tumor sample
In order to create the tumorsbatchscript to do the BAM/SAM processing for thesamtoolspipelinesbatchsubmission script to process the BAM/SAM file usingsamtools, we will once again usesed:
sed 's/normal/tumor/g' samtools_processing_normal.sbatch > samtools_processing_tumor.sbatch
The finalsbatchsubmission script for the tumor sample should look like:#!/bin/bash # This sbatch script is for processing the alignment output from bwa and preparing it for use in GATK using Samtools
# Assign sbatch directives #SBATCH -p priority #SBATCH -t 0-04:00:00 #SBATCH -c 8 #SBATCH --mem 16G #SBATCH -o samtools_processing_tumor_%j.out #SBATCH -e samtools_processing_tumor_%j.err
# Load modules module load gcc/6.2.0 module load samtools/1.15.1
# Assign file paths to variables SAM_FILE=/n/scratch/users/${USER:0:1}/${USER}/variant_calling/alignments/syn3_tumor_GRCh38.p7.sam QUERY_SORTED_BAM_FILE=`echo ${SAM_FILE%sam}query_sorted.bam` FIXMATE_BAM_FILE=`echo ${QUERY_SORTED_BAM_FILE%query_sorted.bam}fixmates.bam` COORDINATE_SORTED_BAM_FILE=`echo ${QUERY_SORTED_BAM_FILE%query_sorted.bam}coordinate_sorted.bam` REMOVED_DUPLICATES_BAM_FILE=`echo ${QUERY_SORTED_BAM_FILE%query_sorted.bam}removed_duplicates.bam`
# Sort SAM file and convert it to a query name sorted BAM file samtools sort \ -@ 8 \ -n \ -o $QUERY_SORTED_BAM_FILE \ $SAM_FILE
# Score mates samtools fixmate \ -@ 8 \ -m \ $QUERY_SORTED_BAM_FILE \ $FIXMATE_BAM_FILE
# Sort BAM file by coordinate samtools sort \ -@ 8 \ -o $COORDINATE_SORTED_BAM_FILE \ $FIXMATE_BAM_FILE
# Mark and remove duplicates and then index the output file samtools markdup \ -r \ --write-index \ -@ 8 \ $COORDINATE_SORTED_BAM_FILE \ ${REMOVED_DUPLICATES_BAM_FILE}##idx##${REMOVED_DUPLICATES_BAM_FILE}.bai
Exercises
1. When inspecting a SAM file you see the following order:
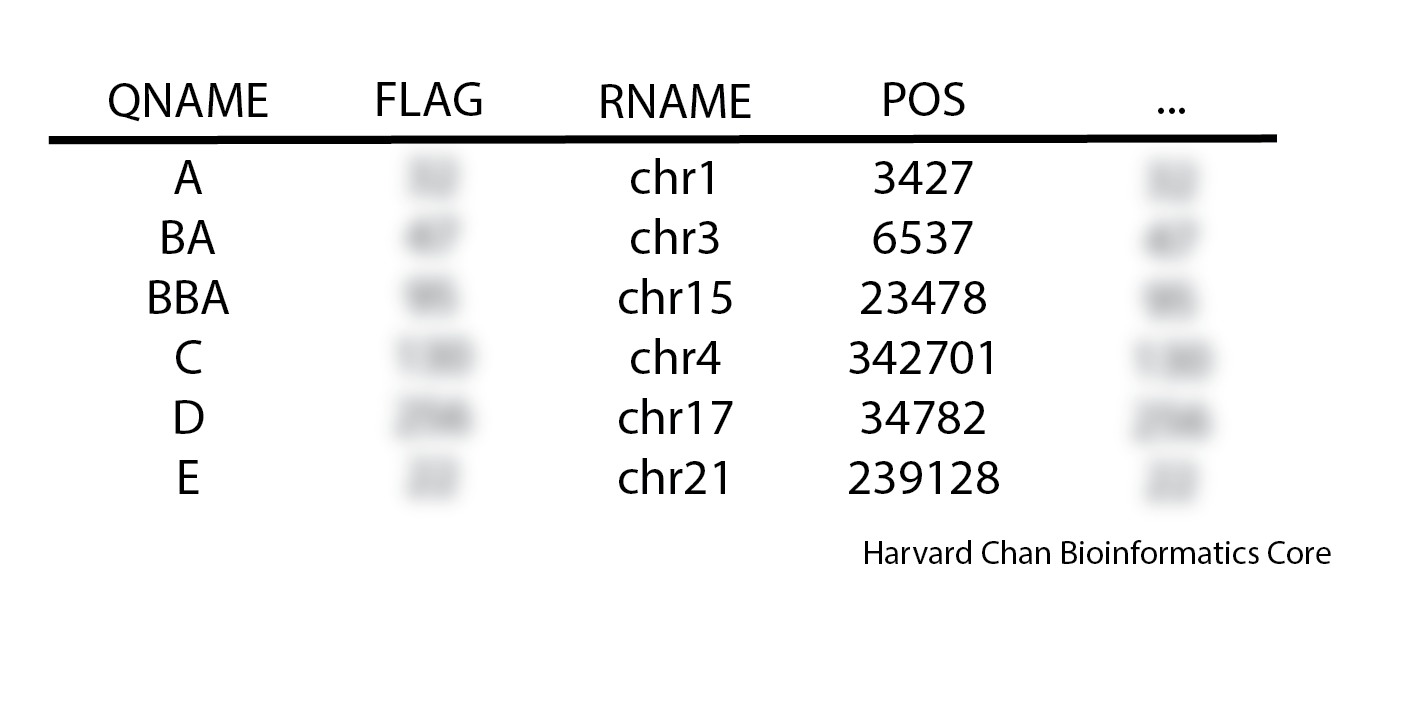
Is this SAM file’s sort order likely: unsorted, query-sorted, coordinate-sorted or is it ambiguous?
Creating the Tumor SAM/BAM processing script
Similar to the bwa script, we will now need use sed to create a sbatch script that will be used for processing the tumor SAM file into a BAM file that can be used as input to GATK. The sed command to do this would be:
sed 's/normal/tumor/g' picard_alignment_processing_normal.sbatch > picard_alignment_processing_tumor.sbatch
As a result your tumor Picard alignment processing script should look almost identical but normal has been replaced by tumor.
Click here to see what our final sbatchcode script for the tumor sample should look like
#!/bin/bash # This sbatch script is for processing the alignment output from bwa and preparing it for use in GATK using Picard
# Assign sbatch directives #SBATCH -p priority #SBATCH -t 0-04:00:00 #SBATCH -c 1 #SBATCH --mem 32G #SBATCH -o picard_alignment_processing_tumor_%j.out #SBATCH -e picard_alignment_processing_tumor_%j.err
# Load module module load picard/2.27.5
# Assign file paths to variables SAMPLE_NAME=syn3_tumor SAM_FILE=/n/scratch/users/${USER:0:1}/${USER}/variant_calling/alignments/${SAMPLE_NAME}_GRCh38.p7.sam REPORTS_DIRECTORY=/home/${USER}/variant_calling/reports/picard/${SAMPLE_NAME}/ QUERY_SORTED_BAM_FILE=`echo ${SAM_FILE%sam}query_sorted.bam` REMOVE_DUPLICATES_BAM_FILE=`echo ${SAM_FILE%sam}remove_duplicates.bam` METRICS_FILE=${REPORTS_DIRECTORY}/${SAMPLE_NAME}.remove_duplicates_metrics.txt COORDINATE_SORTED_BAM_FILE=`echo ${SAM_FILE%sam}coordinate_sorted.bam`
# Make reports directory mkdir -p $REPORTS_DIRECTORY
# Query-sort alginment file and convert to BAM java -jar $PICARD/picard.jar SortSam \ --INPUT $SAM_FILE \ --OUTPUT $QUERY_SORTED_BAM_FILE \ --SORT_ORDER queryname
# Mark and remove duplicates java -jar $PICARD/picard.jar MarkDuplicates \ --INPUT $QUERY_SORTED_BAM_FILE \ --OUTPUT $REMOVE_DUPLICATES_BAM_FILE \ --METRICS_FILE $METRICS_FILE \ --REMOVE_DUPLICATES true
# Coordinate-sort BAM file and create BAM index file java -jar $PICARD/picard.jar SortSam \ --INPUT $REMOVE_DUPLICATES_BAM_FILE \ --OUTPUT $COORDINATE_SORTED_BAM_FILE \ --SORT_ORDER coordinate \ --CREATE_INDEX true
Submitting scripts for Picard processing
Now we are ready to submit our normal and tumor Picard processing scripts to the O2 cluster. However, we might have a problem. If you managed to go quickly into this lesson from the previous lesson, your bwa alignment scripts may still be running and your SAM files are not complete yet!
First, we need to check the status of our bwa scripts and we can do this with the command:
squeue --me
-
If you have
bwajobs still running, then wait for them to complete (less than 2 hours) before continuing. There are ways to queue jobs together in SLURM using the--dependencyoption insbatch. This is outside the scope of this workshop, but we cover this in the automation lesson, if you are interested in learning more. For now, just hang tight until the alignment is complete. -
If the only job running is your interactive job, then it should be time to start your
Picardprocessing scripts. You can go ahead and submit yoursbatchscripts forPicardprocessing with:
sbatch picard_alignment_processing_normal.sbatch
sbatch picard_alignment_processing_tumor.sbatch
NOTE: These scripts will take ~2 hours for each to run!
This lesson has been developed by members of the teaching team at the Harvard Chan Bioinformatics Core (HBC). These are open access materials distributed under the terms of the Creative Commons Attribution license (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.