Approximate time: 90 minutes
Learning Objectives:
- Understand how to bring in data from single-cell RNA-seq experiments
Single-cell RNA-seq: Quality control set-up
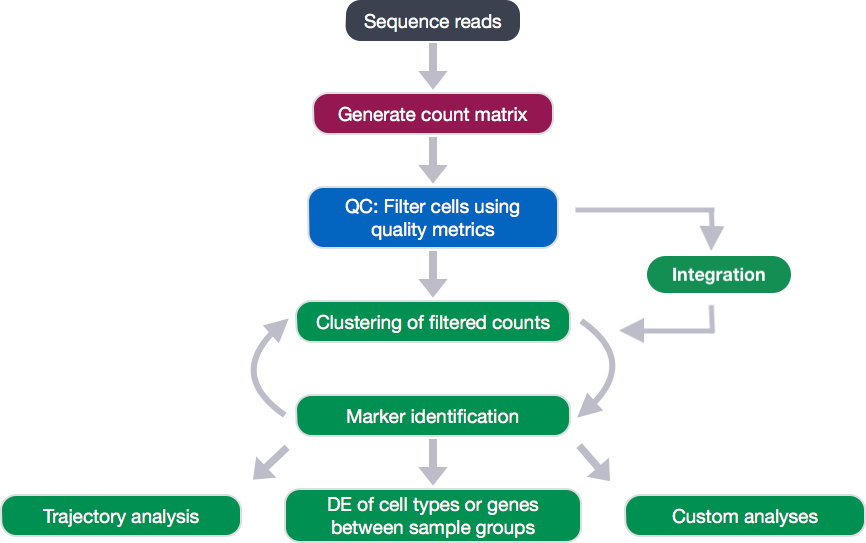
After quantifying gene expression we need to bring this data into R to generate metrics for performing QC. In this lesson we will talk about the format(s) count data can be expected in, and how to read it into R so we can move on to the QC step in the workflow. We will also discuss the dataset we will be using and the associated metadata.
Exploring the example dataset
For this workshop we will be working with a single-cell RNA-seq dataset which is part of a larger study from Kang et al, 2017. In this paper, the authors present a a computational algorithm that harnesses genetic variation (eQTL) to determine the genetic identity of each droplet containing a single cell (singlet) and identify droplets containing two cells from different individuals (doublets).
The data used to test their algorithm is comprised of pooled Peripheral Blood Mononuclear Cells (PBMCs) taken from eight lupus patients, split into control and interferon beta-treated (stimulated) conditions.
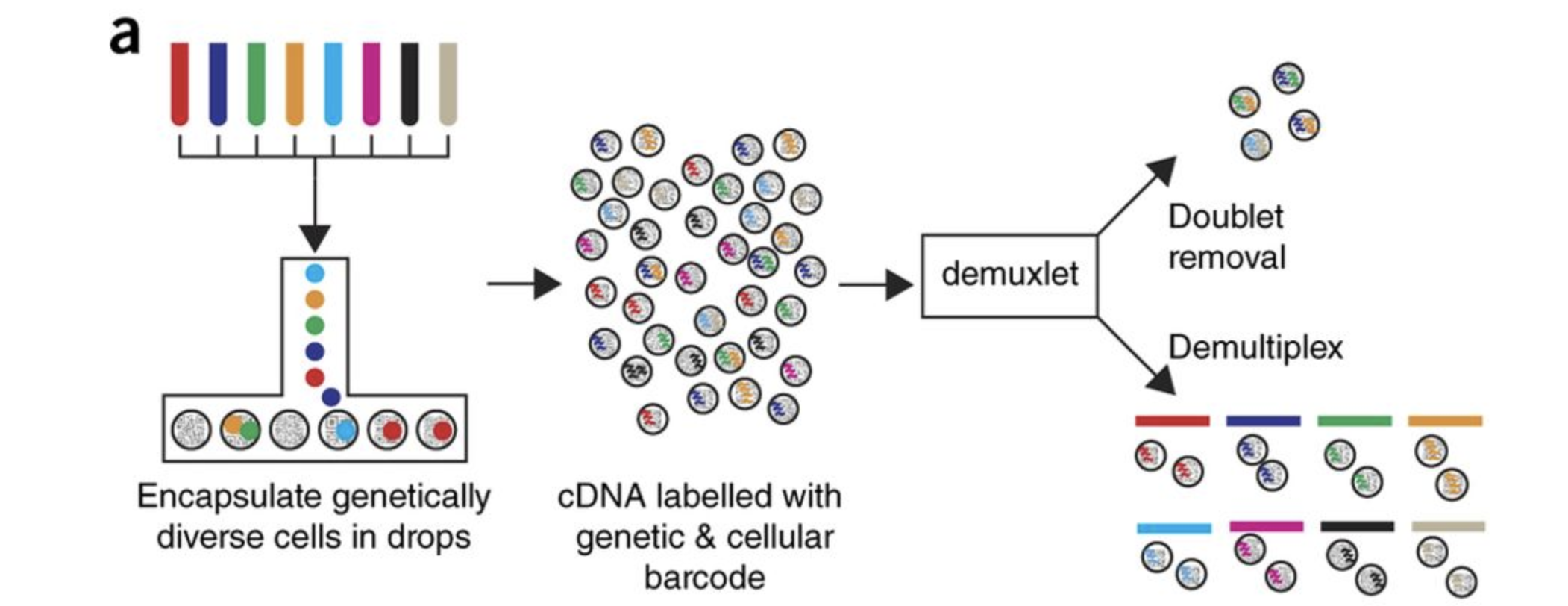
Image credit: Kang et al, 2017
Raw data
This dataset is available on GEO (GSE96583), however the available counts matrix lacked mitochondrial reads, so we downloaded the BAM files from the SRA (SRP102802). These BAM files were converted back to FASTQ files, then run through Cell Ranger to obtain the count data that we will be using.
NOTE: The counts for this dataset is also freely available from 10X Genomics and is used as part of the Seurat tutorial.
Metadata
In addition to the raw data, we also need to collect information about the data; this is known as metadata. There is often a temptation to just start exploring the data, but it is not very meaningful if we know nothing about the samples that this data originated from.
Some relevant metadata for our dataset is provided below:
- The libraries were prepared using 10X Genomics version 2 chemistry
- The samples were sequenced on the Illumina NextSeq 500
- PBMC samples from eight individual lupus patients were separated into two aliquots each.
- One aliquot of PBMCs was activated by 100 U/mL of recombinant IFN-β for 6 hours.
- The second aliquot was left untreated.
- After 6 hours, the eight samples for each condition were pooled together in two final pools (stimulated cells and control cells). We will be working with these two, pooled samples.
-
12,138 and 12,167 cells were identified (after removing doublets) for control and stimulated pooled samples, respectively.
- Since the samples are PBMCs, we will expect immune cells, such as:
- B cells
- T cells
- NK cells
- monocytes
- macrophages
- possibly megakaryocytes
It is recommended that you have some expectation regarding the cell types you expect to see in a dataset prior to performing the QC. This will inform you if you have any cell types with low complexity (lots of transcripts from a few genes) or cells with higher levels of mitochondrial expression. This will enable us to account for these biological factors during the analysis workflow.
None of the above cell types are expected to be low complexity or anticipated to have high mitochondrial content.
Setting up the R environment
One of the most important parts of research that involves large amounts of data, is how best to manage it. We tend to prioritize the analysis, but there are many other important aspects of data management that are often overlooked in the excitement to get a first look at new data. The HMS Data Management Working Group, discusses in-depth some things to consider beyond the data creation and analysis.
One important aspect of data management is organization. For each experiment you work on and analyze data for, it is considered best practice to get organized by creating a planned storage space (directory structure). We will do that for our single-cell analysis.
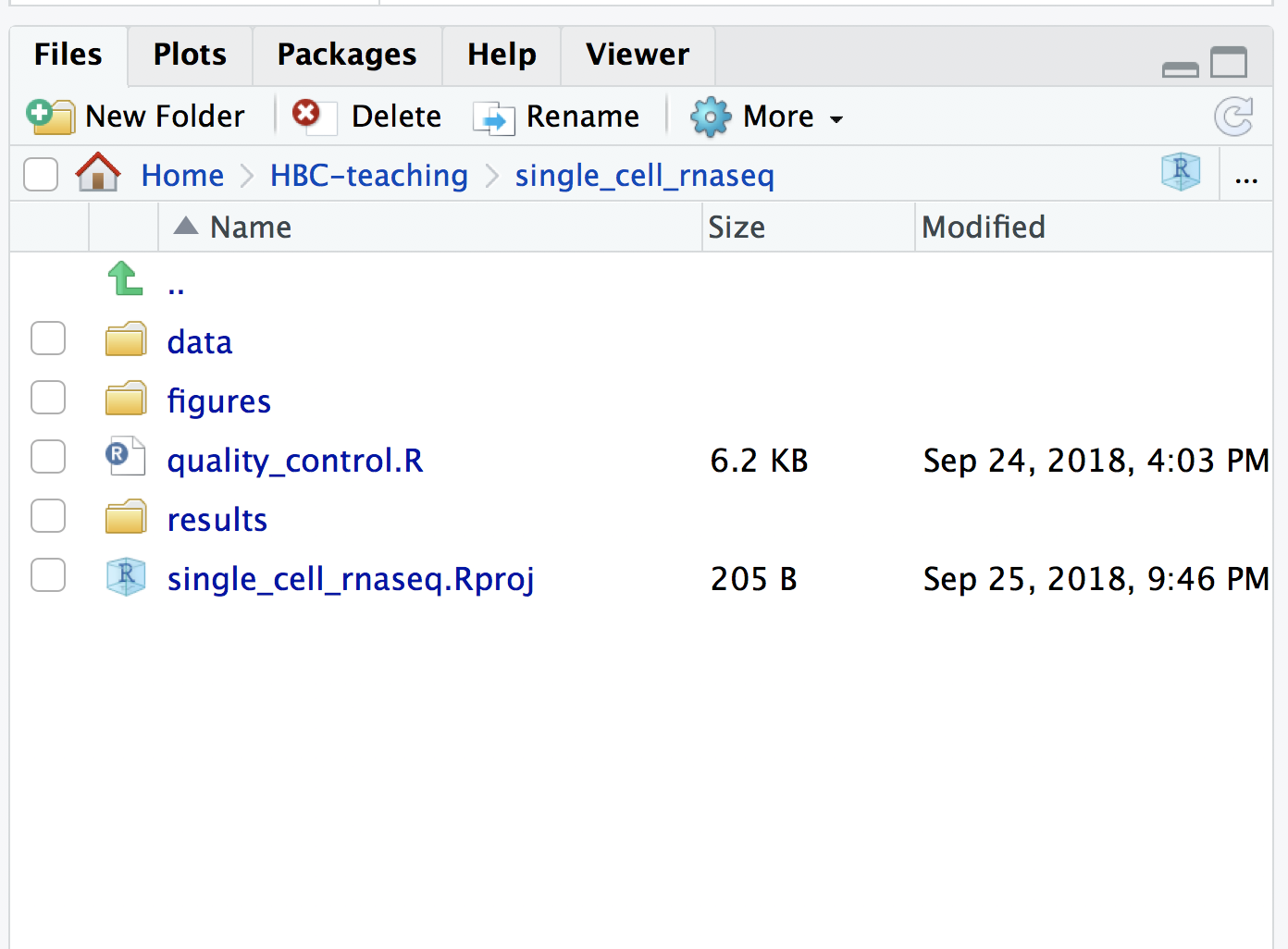
Open up RStudio and create a new R project entitled single_cell_rnaseq. Then, create the following directories:
single_cell_rnaseq/
├── data
├── results
└── figures
Download data
Right-click the links below to download the output folders from Cell Ranger for each sample into the data folder:
Now, let’s unzip the two zipped folders that we just downloaded by double-clicking, so we can see their contents from within RStudio.
New script
Next, open a new Rscript file, and start with some comments to indicate what this file is going to contain:
# February 2020
# HBC single-cell RNA-seq workshop
# Single-cell RNA-seq analysis - QC
Save the Rscript as quality_control.R. Your working directory should look something like this:
Loading libraries
Now, we can load the necessary libraries:
# Load libraries
library(SingleCellExperiment)
library(Seurat)
library(tidyverse)
library(Matrix)
library(scales)
library(cowplot)
library(RCurl)
Loading single-cell RNA-seq count data
Regardless of the technology or pipeline used to process your single-cell RNA-seq sequence data, the output will generally be the same. That is, for each individual sample you will have the following three files:
- a file with the cell IDs, representing all cells quantified
- a file with the gene IDs, representing all genes quantified
- a matrix of counts per gene for every cell
We can explore these files by clicking on the data/ctrl_raw_feature_bc_matrix folder:
1. barcodes.tsv
This is a text file which contains all cellular barcodes present for that sample. Barcodes are listed in the order of data presented in the matrix file (i.e. these are the column names).

2. features.tsv
This is a text file which contains the identifiers of the quantified genes. The source of the identifier can vary depending on what reference (i.e. Ensembl, NCBI, UCSC) you use in the quantification methods, but most often these are official gene symbols. The order of these genes corresponds to the order of the rows in the matrix file (i.e. these are the row names).
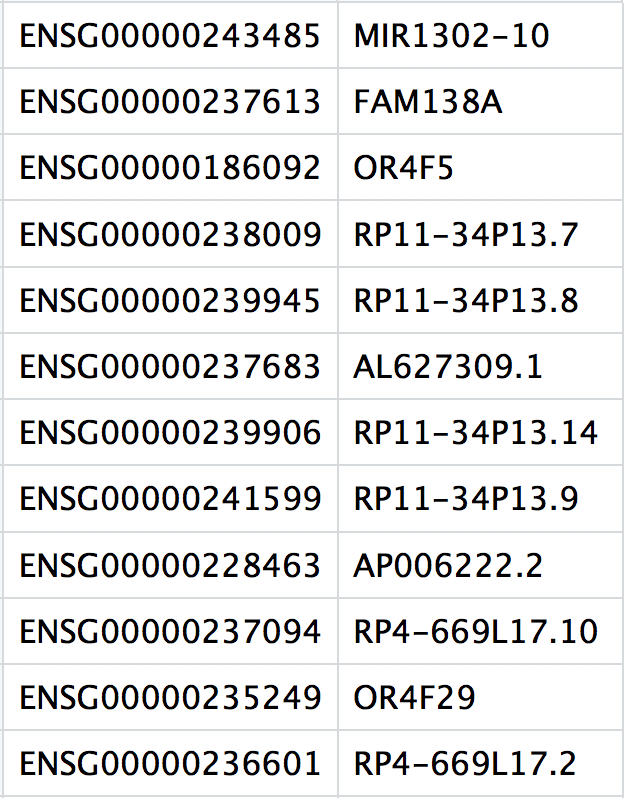
3. matrix.mtx
This is a text file which contains a matrix of count values. The rows are associated with the gene IDs above and columns correspond to the cellular barcodes. Note that there are many zero values in this matrix.
Loading this data into R requires us to use functions that allow us to efficiently combine these three files into a single count matrix. However, instead of creating a regular matrix data structure, the functions we will use create a sparse matrix to improve the amount of space, memory and CPU required to work with our huge count matrix.
Different methods for reading in data include:
readMM(): This function is from the Matrix package and will turn our standard matrix into a sparse matrix. Thefeatures.tsvfile andbarcodes.tsvmust first be individually loaded into R and then they are combined. For specific code and instructions on how to do this please see our additional material.Read10X(): This function is from the Seurat package and will use the Cell Ranger output directory as input. In this way individual files do not need to be loaded in, instead the function will load and combine them into a sparse matrix for you. We will be using this function to load in our data!
Reading in a single sample (read10X())
When working with 10X data and its proprietary software Cell Ranger, you will always have an outs directory. Within this directory you will find a number of different files including:
web_summary.html: report that explores different QC metrics, including the mapping metrics, filtering thresholds, estimated number of cells after filtering, and information on the number of reads and genes per cell after filtering.- BAM alignment files: files used for visualization of the mapped reads and for re-creation of FASTQ files, if needed
filtered_feature_bc_matrix: folder containing all files needed to construct the count matrix using data filtered by Cell Rangerraw_feature_bc_matrix: folder containing all files needed to construct the count matrix using the raw unfiltered data
We are mainly interested in the raw_feature_bc_matrix as we wish to perform our own QC and filtering while accounting for the biology of our experiment/biological system.
If we had a single sample, we could generate the count matrix and then subsequently create a Seurat object:
# How to read in 10X data for a single sample (output is a sparse matrix)
ctrl_counts <- Read10X(data.dir = "data/ctrl_raw_feature_bc_matrix")
# Turn count matrix into a Seurat object (output is a Seurat object)
ctrl <- CreateSeuratObject(counts = ctrl_counts,
min.features = 100)
NOTE: The
min.featuresargument specifies the minimum number of genes that need to be detected per cell. This argument will filter out poor quality cells that likely just have random barcodes encapsulated without any cell present. Usually, cells with less than 100 genes detected are not considered for analysis.
Seurat automatically creates some metadata for each of the cells when you use the Read10X() function to read in data. This information is stored in the meta.data slot within the Seurat object (see more in the note below).
The Seurat object is a custom list-like object that has well-defined spaces to store specific information/data. You can find more information about the slots in the Seurat object at this link.
# Explore the metadata
head(ctrl@meta.data)
What do the columns of metadata mean?
orig.ident: this often contains the sample identity if known, but will default to “SeuratProject”nCount_RNA: number of UMIs per cellnFeature_RNA: number of genes detected per cell
Reading in multiple samples with a for loop
In practice, you will likely have several samples that you will need to read in data for using one of the 2 functions we discussed earlier (Read10X() or readMM()). So, to make the data import into R more efficient we can use a for loop, that will interate over a series of commands for each of the inputs given.
In R, it has the following structure/syntax:
## DO NOT RUN
for (variable in input){
command1
command2
command3
}
The for loop we will be using today will iterate over the two sample “files” and execute two commands for each sample - (1) read in the count data (Read10X()) and (2) create the Seurat objects from the read in data (CreateSeuratObject()):
# Create each individual Seurat object for every sample
for (file in c("ctrl_raw_feature_bc_matrix", "stim_raw_feature_bc_matrix")){
seurat_data <- Read10X(data.dir = paste0("data/", file))
seurat_obj <- CreateSeuratObject(counts = seurat_data,
min.features = 100,
project = file)
assign(file, seurat_obj)
}
We can break down the
for loopto describe the different lines of code:Step 1: Specify inputs
For this dataset, we have two samples that we would like to create a Seurat object for:
ctrl_raw_feature_bc_matrixstim_raw_feature_bc_matrixWe can specify these samples in the input part for our
for loopas elements of a vector usingc(). We are assigning these to a variable and we can call that variable anything we would like (try to give it a name that makes sense). In this example, we called the variablefile.During the execution of the above loop,
filewill first contain the value “ctrl_raw_feature_bc_matrix”, run through the commands all the way through toassign(). Next, it will contain the value “stim_raw_feature_bc_matrix” and once again run through all the commands. If you had 15 folders as input, instead of 2, the above code will run through 15 times, for each of your data folders.## DO NOT RUN # Create each individual Seurat object for (file in c("ctrl_raw_feature_bc_matrix", "stim_raw_feature_bc_matrix")){Step 2: Read in data for the input
We can continue our
for loopby adding a line to read in data withRead10X():
- Here, we need to specify the path to the file, so we will prepend the
data/directory to our sample folder name using thepaste0()function.## DO NOT RUN seurat_data <- Read10X(data.dir = paste0("data/", file))Step 3: Create Seurat object from the 10X count data
Now, we can create the Seurat object by using the
CreateSeuratObject()function, adding in the argumentproject, where we can add the sample name.## DO NOT RUN seurat_obj <- CreateSeuratObject(counts = seurat_data, min.features = 100, project = file)Step 4: Assign Seurat object to a new variable based on sample
The last command
assigns the Seurat object created (seurat_obj) to a new variable. In this way, when we iterate and move on to the next sample in ourinputwe will not overwrite the Seurat object created in the previous iteration:## DO NOT RUN assign(file, seurat_obj) }
Now that we have created both of these objects, let’s take a quick look at the metadata to see how it looks:
# Check the metadata in the new Seurat objects
head(ctrl_raw_feature_bc_matrix@meta.data)
head(stim_raw_feature_bc_matrix@meta.data)
Next, we need to merge these objects together into a single Seurat object. This will make it easier to run the QC steps for both sample groups together and enable us to easily compare the data quality for all the samples.
We can use the merge() function from the Seurat package to do this:
# Create a merged Seurat object
merged_seurat <- merge(x = ctrl_raw_feature_bc_matrix,
y = stim_raw_feature_bc_matrix,
add.cell.id = c("ctrl", "stim"))
Because the same cell IDs can be used for different samples, we add a sample-specific prefix to each of our cell IDs using the add.cell.id argument. If we look at the metadata of the merged object we should be able to see the prefixes in the rownames:
# Check that the merged object has the appropriate sample-specific prefixes
head(merged_seurat@meta.data)
tail(merged_seurat@meta.data)
This lesson has been developed by members of the teaching team at the Harvard Chan Bioinformatics Core (HBC). These are open access materials distributed under the terms of the Creative Commons Attribution license (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.