Approximate time: 45 minutes
Learning Objectives
- Differentiate between germline and somatic variant calling
- Call somatic variants from
bamfiles usingMuTect2
Variant Calling
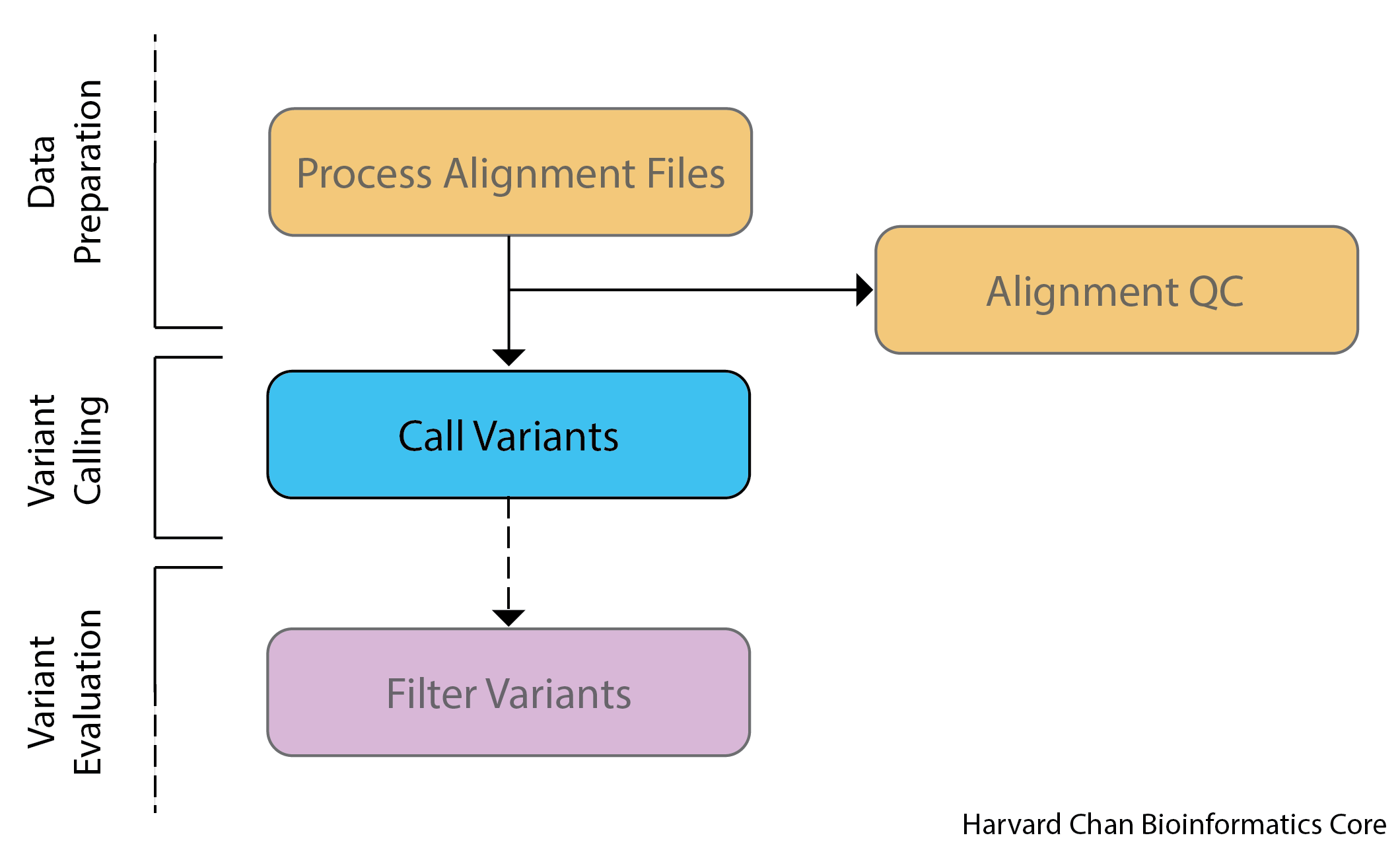
Best practices for variant calling
Now that we have inspected our reads and alignments for high-quality QC and also formatted our alignments, we are ready for variant calling. Dozens of variant calling tools for NGS data have been published, and choosing which one to use can be a daunting task. Accurate variant detection in the human genome is a key requirement for diagnostics in clinical sequencing, and so you want to make sure you are using the caller which is appropriate for your data and study design.
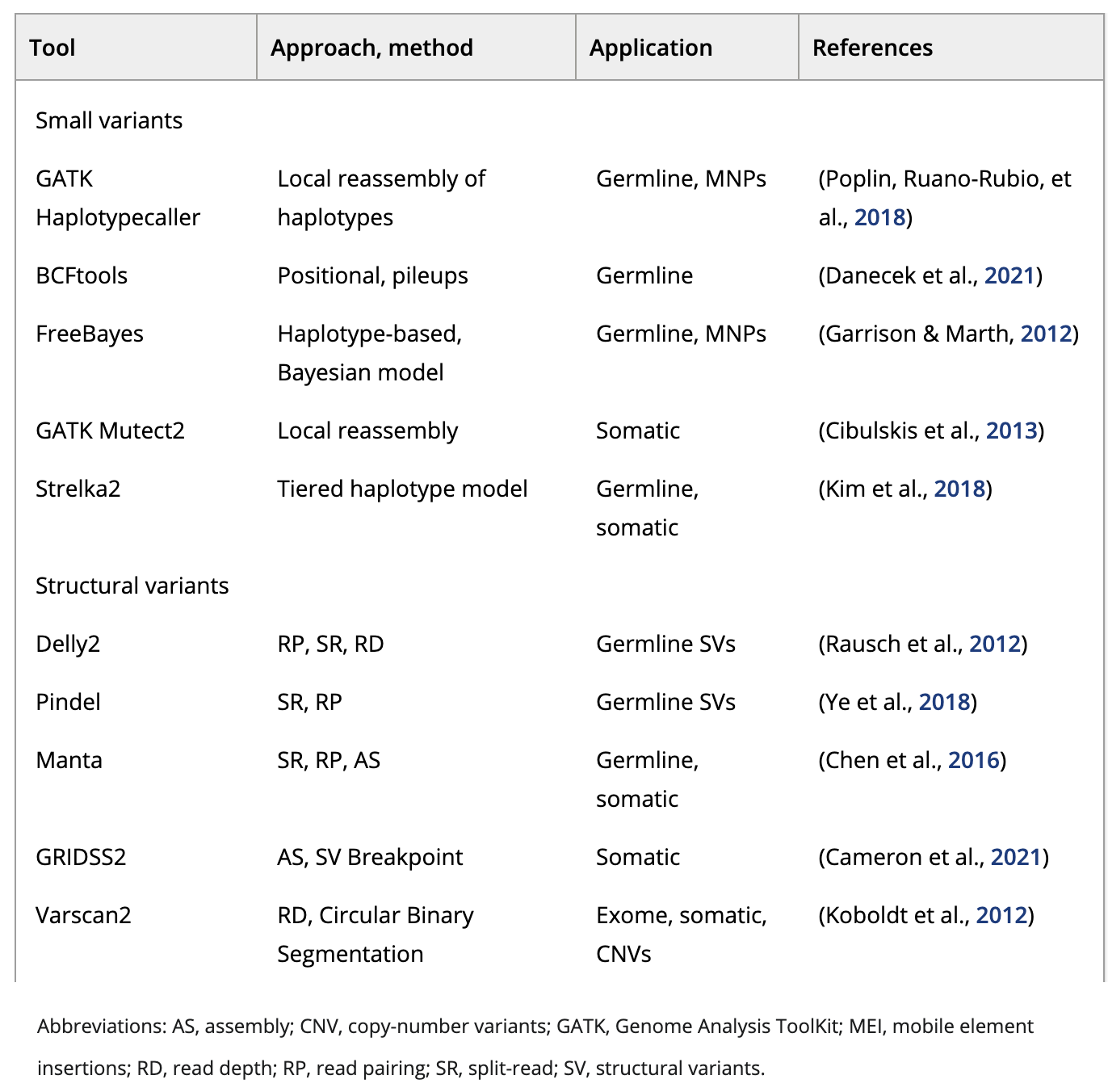
Image source: Zverinova S and Guryev V, 2021
Germline versus Somatic Variant Calling
Variant calling can be broadly broken up into two groups, germline and somatic. These two types of variant calling methods have different assumptions regarding the input data and thus are handled differently.
-
Germline variant calling refers to the process of calling variants that are ubiquitous across the organism (i.e. almost all cells carry these variants) and these are the types of variants that can be passed through the germline. Germline variant calling for the most part expects at most two alleles in relatively equal frequencies.
-
Somatic variant calling refers to the process of calling variants that differ between cells within a single organism and these variants are not passed through the germline. Somatic variant calling is often used when studying the progression of various cancers. Somatic variant calling is more difficult than germline variant calling because low frequency variants and sequencing artifacts are difficult to distinguish from sequencing errors.
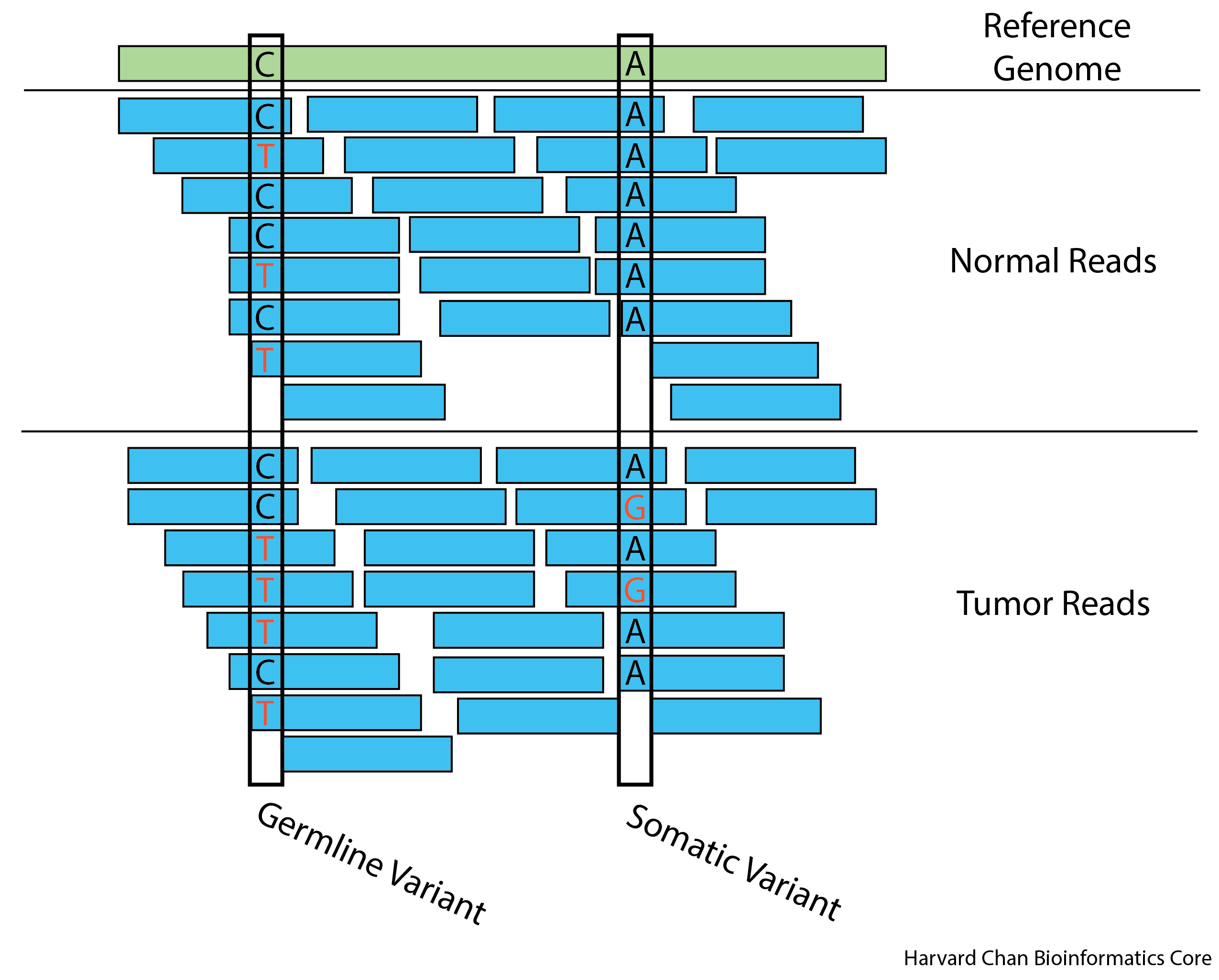
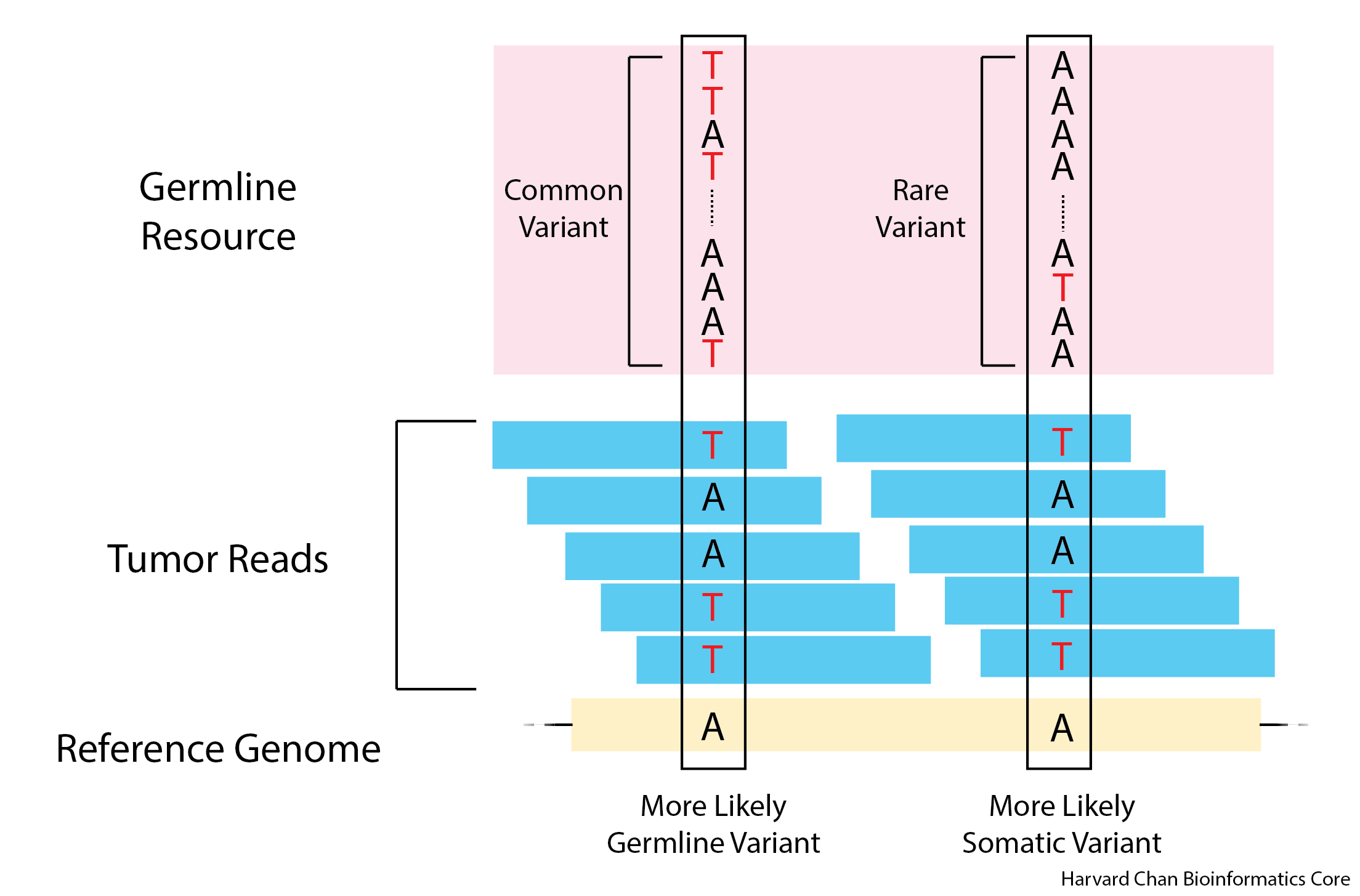
In the above image we can see an example of a germline variant on the left. Approximately half of the reads support each allele in both the tumor and normal sample reads. This is compared to the somatic variant on the right, where we observe no variants in the normal sample reads but there is a variant present in the tumor sample reads.
For this workshop, we have tumor/normal samples and so we will be calling somatic variants.
Panel of Normals
A panel of normals (PoN) is a resource derived from a set of healthy tissues with the goal of correcting for recurrent technical artifacts within a sequencing experiment. Ideally, a panel of normals should:
- Include samples from healthy individuals, which can be unrelated to the samples in the analysis
- Preferably include younger individuals, to reduce the noise of somatic mutations or undiagnosed tumors
- Undergo the same sample preparation and sequencing as the samples of interest
- Include at least 40 individuals
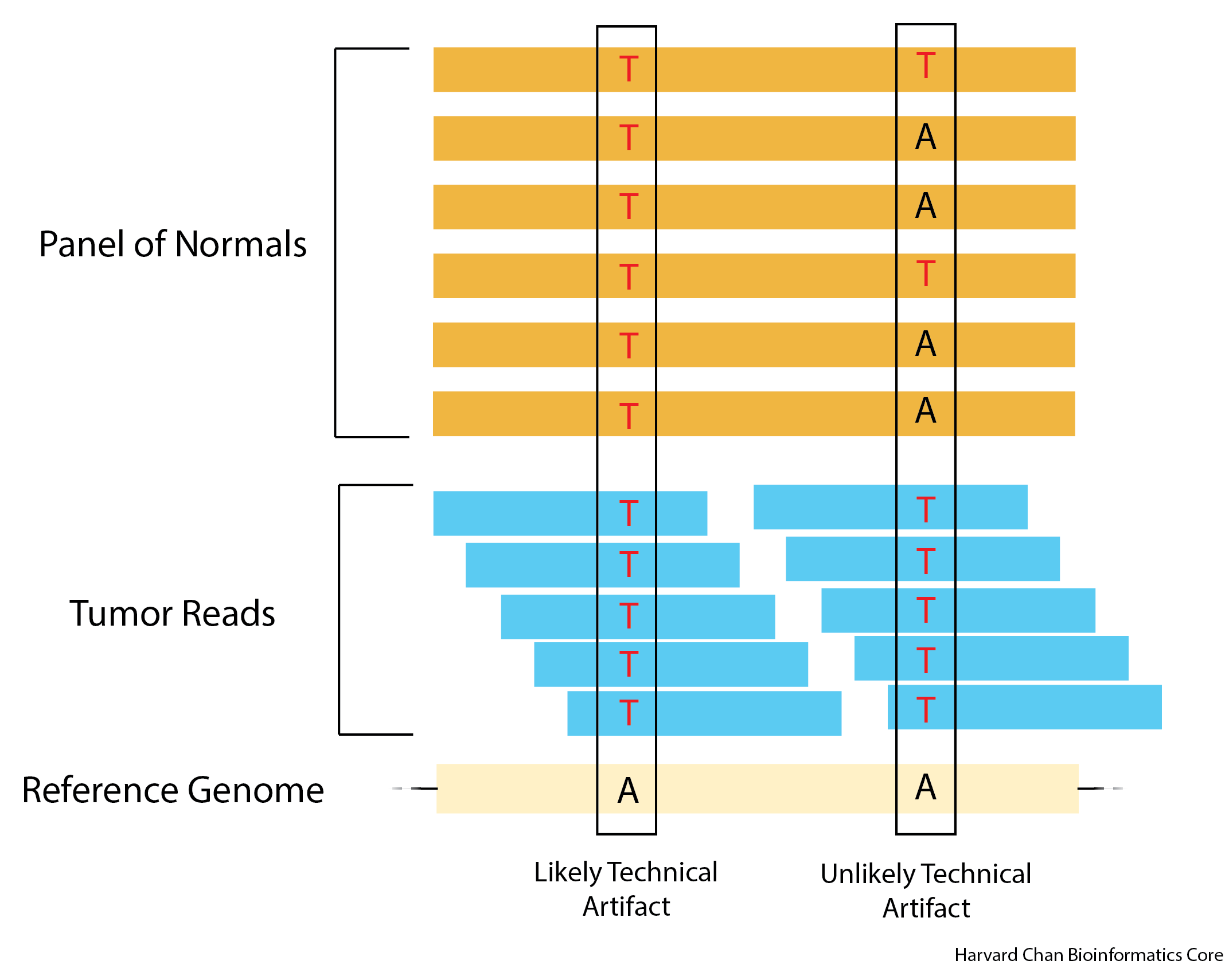
In the above image we can see that for the left “variant”, the panel of normals and the tumor all have a thymine “variant” in this position relative to the reference. This would likely be a technical artifact. However, for the right variant, we can see that while the tumor thymine allele is still present in all of the reads for this position like the previous case, the panel of normals shows variation and this signifies that this position is likely not a technical artifact.
An additional benefit of a good panel of normals is that it can act as an additional filter for germline variants. This is particularly true when you run MuTect2 in tumor-only mode (discussed briefly later). If you do not have access to a panel of normals, you can also use a panel of normals that the Broad Institute hosts which is derived from the 1000 Genomes Project. This data can be downloaded here. We will are currently testing the best way to implement this and will hopefully have a implementation next week. The Broad Institute also provides instructions on how to make your own panel of normals here. It is not required to use a panel of normals when running Mutect2, but it is recommended.
Note: When using a panel of normals VCF file in MuTect2, you will also need an index of the VCF file. We have already created the index for this workshop, but this can be accomplished in one of a few ways:
- If you are using the panel of normals provided by the Broad Institute, then they also provide a index that you can download from here.
- You can index your own VCF file with the
IndexFeatureFilewithin GATK.- You can also use other software packages like tabix or bcftools to create index files for VCF files
Germline Resource
A germline resource contains many variants within a population and can be used in conjunction with other tools to help variant calling algorithms determine if a variant is likely a germline or somatic mutation. The logic is: if a variant is a common variant in the population and it is present in the sample, then it is more likely to be a germline variant rather than a somatic variant. On the other hand, if a low-frequency variant in the population is present in an sample, then it is more likely to be somatic variant rather than a germline variant. This idea is illustrated below:
The Broad Institute provides a VCF file utilizing the variants from gnomAD, which can be used as a germline reference. Similarly to a panel of normals, this is not required for MuTect2 to call variants, however, it is also recommended. We will are currently testing the best way to implement this and will hopefully have a implementation next week.
Note: Similarly to the index file needed for the panel of normals VCF file, we will also need a index file for the germline resource VCF file. If you are using the Broad Institute’s gnomAD VCF file, then you can download the index from here or create it yourself using GATK, tabix or bcftools (discussed in the note above).
GATK Toolkit
GATK (Genome Analysis Toolkit) is a popular open-source software package developed by the Broad Institute for analysis of high throughput sequencing (HTS) data. It was initially developed around 2007 to help analyze sequencing data from the 1000 Genomes Project. It was created with a focus on best practices in variant discovery and genotyping from NGS data. New versions of GATK are regularly released by the Broad Institute as sequencing technologies and reference genomes are improved.
![]()
GATK has tools for preprocessing, variant discovery, genotyping, and filtering of HTS data. It takes aligned sequencing reads as input and outputs variant calls. There are several different GATK Best Practices workflows tailored to particular applications depending on the type of variation of interest and the technology employed.
In terms of variant discovery, GATK offers the following tools:
HaplotypeCallercan be used for germline SNPs and IndelsMuTect2can be used for somatic SNPs and IndelsGermlineCNVCallercan be used for germline Copy Number Variants- Software for Structural Variants is currently in beta testing.
This course is going to focus on analyzing somatic SNPs, so we are going to use MuTect2.
MuTect2
Basic workflow
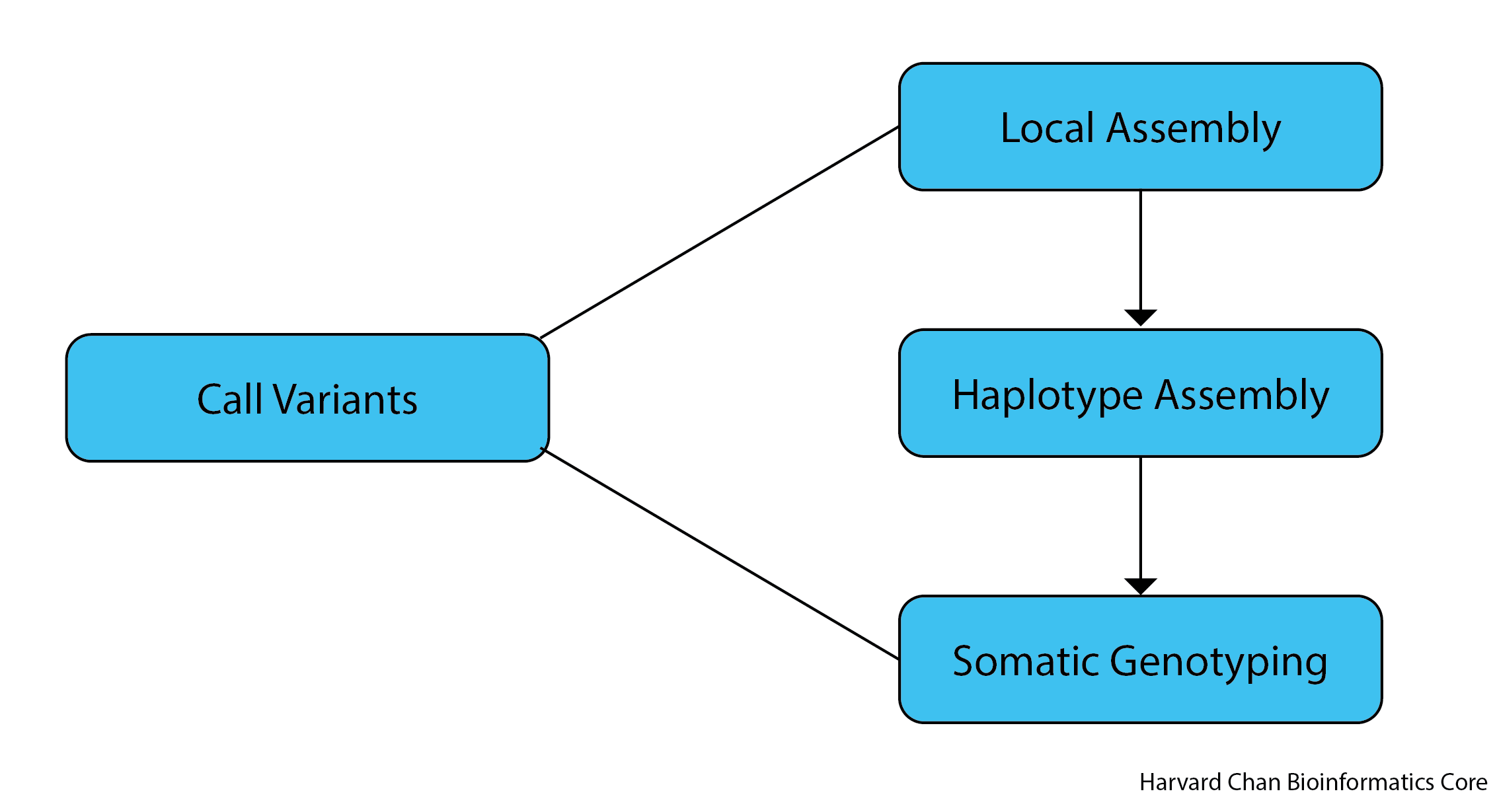
The algorithm used for somatic variant discovery with MuTect2 is broken up into three major components:
- Local Assembly - The initial step is for MuTect2 to evaluates regions for somatic variations. Regions that surpass a log odds threshold for somatic variation comparing the existance and non-existance of a potential alternate allele are flagged as “active” regions and are slated for local reassembly and realignment. The local reassembly is done by creating a de Bruijn graph of the active region.
- Haplotype Assembly - From this de Bruijn graph, each haplotype is assembled and the haplotype spans the variant in question and connects to two points in the reference genome.
- Somatic Genotyping - From here, most likely haplotypes are assembled and variants are called from these haplotypes.
MuTect2 can be run in three different modes:
- Tumor-normal mode: where a tumor sample is matched with a normal samples. This is the mode we will be using.
- Tumor-only mode: where a single sample is used as input.
- Note that the software’s ability to reliably call somatic variants is greatly diminished as it has difficulty distinguishing between high-frequency variants and germline variants. In order to help, a panel of normals and a germline resource are strongly recommended when running in this mode.
- Mitochondrial mode: where specific parameters are added for appropriately calling on mitochondria.
Running MuTect2
Let’s start our variant calling analysis by writing out a new sbatch submission script for MuTect2:
cd ~/variant_calling/scripts/
vim mutect2_normal_tumor.sbatch
Let’s add our shebang line, script description and sbatch directives:
#!/bin/bash
# This sbatch script is for variant calling with GATK's MuTect2
# Assign sbatch directives
#SBATCH -p priority
#SBATCH -t 1-00:00:00
#SBATCH -c 1
#SBATCH --mem 16G
#SBATCH -o mutect2_variant_calling_normal_tumor_%j.out
#SBATCH -e mutect2_variant_calling_normal_tumor_%j.err
Next to add the GATK4 module we are going to load:
# Load the GATK module
module load gatk/4.1.9.0
And now, we need to create our variables:
# Assign variables
REFERENCE_SEQUENCE=/n/groups/hbctraining/variant_calling/reference/GRCh38.p7.fa
REFERENCE_DICTIONARY=`echo ${REFERENCE_SEQUENCE%fa}dict`
NORMAL_SAMPLE_NAME=syn3_normal
NORMAL_BAM_FILE=/n/scratch/users/${USER:0:1}/${USER}/variant_calling/alignments/${NORMAL_SAMPLE_NAME}_GRCh38.p7.coordinate_sorted.bam
TUMOR_SAMPLE_NAME=syn3_tumor
TUMOR_BAM_FILE=/n/scratch/users/${USER:0:1}/${USER}/variant_calling/alignments/${TUMOR_SAMPLE_NAME}_GRCh38.p7.coordinate_sorted.bam
VCF_OUTPUT_FILE=/n/scratch/users/${USER:0:1}/${USER}/variant_calling/vcf_files/mutect2_${NORMAL_SAMPLE_NAME}_${TUMOR_SAMPLE_NAME}_GRCh38.p7-raw.vcf
Click here if you used samtools instead of Picard to process the alignment files
Very little needs to be edited to these variables to adapt them for the samtools output. However, the end of the file that was created in samtools was .removed_duplicates.bam rather than .coordinate_sorted.bam. As a result we need to edit the variables a bit. Change:
NORMAL_BAM_FILE=/n/scratch/users/${USER:0:1}/${USER}/variant_calling/alignments/${NORMAL_SAMPLE_NAME}_GRCh38.p7.coordinate_sorted.bam
To:
NORMAL_BAM_FILE=/n/scratch/users/${USER:0:1}/${USER}/variant_calling/alignments/${NORMAL_SAMPLE_NAME}_GRCh38.p7.removed_duplicates.bam
And also change:
TUMOR_BAM_FILE=/n/scratch/users/${USER:0:1}/${USER}/variant_calling/alignments/${TUMOR_SAMPLE_NAME}_GRCh38.p7.coordinate_sorted.bam
To:
TUMOR_BAM_FILE=/n/scratch/users/${USER:0:1}/${USER}/variant_calling/alignments/${TUMOR_SAMPLE_NAME}_GRCh38.p7.removed_duplicates.bam
After those changeed have been made, the rest of the script should be the same.
NOTE: Sometimes when there are many input variables that re-use many of the same textual elements (i.e. paths, sample names and reference genome names), like we have above, it is sometimes cleaner, less typo-prone and more reproducible to assign those repeated items to variables and then use text manipulation tools and variable subsitution in
bashto create the rest of the variables. In the above example, some of the lines of variable assignment (REFERENCE_SEQUENCE,NORMAL_SAMPLE_NAMEandTUMOR_SAMPLE_NAME) are likely lines you might edit from run-to-run, but the other lines (REFERENCE_DICTIONARY,NORMAL_BAM_FILE,TUMOR_BAM_FILEandVCF_OUTPUT_FILE) will likely stay the same or similiar. Standardizing your paths and nomenclature will help you keep track of your files much easier.
Lastly, we need to add the MuTect2 command:
# Run MuTect2
gatk Mutect2 \
--sequence-dictionary $REFERENCE_DICTIONARY \
--reference $REFERENCE_SEQUENCE \
--input $NORMAL_BAM_FILE \
--normal-sample $NORMAL_SAMPLE_NAME \
--input $TUMOR_BAM_FILE \
--tumor-sample $TUMOR_SAMPLE_NAME \
--annotation ClippingRankSumTest --annotation DepthPerSampleHC --annotation MappingQualityRankSumTest --annotation MappingQualityZero --annotation QualByDepth --annotation ReadPosRankSumTest --annotation RMSMappingQuality --annotation FisherStrand --annotation MappingQuality --annotation DepthPerAlleleBySample --annotation Coverage \
--output $VCF_OUTPUT_FILE
Let’s breakdown this command:
--sequence-dictionary $REFERENCE_DICTIONARY:GATKrequires a sequence directory (.dict) file of the reference sequence. We have gone ahead and already created this for you.--reference $REFERENCE_SEQUENCE: This is the genome reference sequence.--input $NORMAL_BAM_FILE: This is the firstbamfile that we are providing GATK and it happens to be the normal sample--normal-sample $NORMAL_SAMPLE_NAME: This is the name of that normal sample and will be used as a column header in the VCF file--input $TUMOR_BAM_FILE: This is the secondbamfile that we are providing GATK and it happens to be the tumor sample--tumor-sample $TUMOR_SAMPLE_NAME: This is the name of that tumor sample and will be used as a column header in the VCF file
NOTE: It is VERY IMPORTANT that the sample names (
--normal-sample $NORMAL_SAMPLE_NAMEand--tumor-sample $TUMOR_SAMPLE_NAME) are provided in the same order as the--inputinput BAM files!
- Parameters related to
--annotation: These are a variety of additional annotations that we are going to add to the output VCF file. These are not required forMuTect2to run, but they provide additional details about our variants. --output $VCF_OUTPUT_FILE: This is our output VCF file
In order to run MuTect2 we also need to have a FASTA index file of our reference sequence in addition to our sequence dictionary. Similarly to the sequence dictionary, VCF indices and bwa indicies, we have already created this index for you. However, we have two dropdowns below to walk you through how to do it, should you ever need to do it on your own.
Click here for details for creating a FASTA index file in
samtools
FASTA index files for reference sequences are fairly common requirements for a variety of NGS software packages.Picardcurrently does not feature an ability to create a FASTA index file. However,samtoolsis a very popular tool that is used for a variety of processes for processing BAM/SAM files and it also includes functionality for the creation of FASTA index files. First, we will need to load thegccandsamtoolsmodules:module load gcc/6.2.0 module load samtools/1.15.1The command for indexing a FASTA file is straightforward and should run pretty quickly:# YOU DON'T NEED TO RUN THIS samtools faidx \ reference_sequence.faWe can breakdown this code:Once the indexing is complete, then you should have a index file (
samtools faidxThis calls thefaidxsoftware fromsamtoolsreference_sequence.faThis is the reference sequence FASTA file that you would like to indexreference_sequence.fa.fai) in same directory as your reference sequencereference_sequence.fa.
Click here for the commands to create a sequence directory
We can create the required sequence dictionary inPicard. But first, let's double check we have thePicardmodule loaded:module load picard/2.27.5The command to do create the sequence dictionary is:
# YOU DON'T NEED TO RUN THIS java -jar $PICARD/picard.jar CreateSequenceDictionary \ --REFERENCE /n/groups/hbctraining/variant_calling/reference/GRCh38.p7_genomic.fa --OUTPUT /n/groups/hbctraining/variant_calling/reference/GRCh38.p7_genomic.dictThe components of this command are:Like indexing, once you have created the sequence dictionary for a reference genome, you won't need to do it again.
java -jar $PICARD/picard.jar CreateSequenceDictionaryThis calls theCreateSequenceDictionarycommand withinPicard--REFERENCE /n/groups/hbctraining/variant_calling/reference/GRCh38.p7_genomic.faThis is the reference sequence to create the sequence dictionary from.--OUTPUT /n/groups/hbctraining/variant_calling/reference/GRCh38.p7_genomic.dictThis is the output sequence dictionary.
Click here to see what our final sbatchcode script for calling variants with MuTect2 should look like
#!/bin/bash # This sbatch script is for variant calling with GATK's MuTect2
# Assign sbatch directives #SBATCH -p priority #SBATCH -t 1-00:00:00 #SBATCH -c 1 #SBATCH --mem 16G #SBATCH -o mutect2_variant_calling_normal_tumor_%j.out #SBATCH -e mutect2_variant_calling_normal_tumor_%j.err
# Load the GATK module module load gatk/4.1.9.0
# Assign variables REFERENCE_SEQUENCE=/n/groups/hbctraining/variant_calling/reference/GRCh38.p7.fa REFERENCE_DICTIONARY=`echo ${REFERENCE_SEQUENCE%fa}dict` NORMAL_SAMPLE_NAME=syn3_normal NORMAL_BAM_FILE=/n/scratch/users/${USER:0:1}/${USER}/variant_calling/alignments/${NORMAL_SAMPLE_NAME}_GRCh38.p7.coordinate_sorted.bam TUMOR_SAMPLE_NAME=syn3_tumor TUMOR_BAM_FILE=/n/scratch/users/${USER:0:1}/${USER}/variant_calling/alignments/${TUMOR_SAMPLE_NAME}_GRCh38.p7.coordinate_sorted.bam VCF_OUTPUT_FILE=/n/scratch/users/${USER:0:1}/${USER}/variant_calling/vcf_files/mutect2_${NORMAL_SAMPLE_NAME}_${TUMOR_SAMPLE_NAME}_GRCh38.p7-raw.vcf
# Run MuTect2 gatk Mutect2 \ --sequence-dictionary $REFERENCE_DICTIONARY \ --reference $REFERENCE_SEQUENCE \ --input $NORMAL_BAM_FILE \ --normal-sample $NORMAL_SAMPLE_NAME \ --input $TUMOR_BAM_FILE \ --tumor-sample $TUMOR_SAMPLE_NAME \ --annotation ClippingRankSumTest --annotation DepthPerSampleHC --annotation MappingQualityRankSumTest --annotation MappingQualityZero --annotation QualByDepth --annotation ReadPosRankSumTest --annotation RMSMappingQuality --annotation FisherStrand --annotation MappingQuality --annotation DepthPerAlleleBySample --annotation Coverage \ --output $VCF_OUTPUT_FILE
You can submit your variant calling script to the cluster:
sbatch mutect2_normal_tumor.sbatch
VCF (Variant Call Format)
The output from MuTect2 is a VCF file; the de facto file format for storing genetic variation. The Variant Call Format (VCF) is a standardized, text-file format for describing variants identifed from a sequencing experiment. This allows for downstream processes to be streamlined and also allows for researchers to easily collaborate and wrangle a shared set of variant calls.
Since it is a text file, we could easily take a quick peek at our VCF file using the less command. However, our script might still be runnning!
Instead, we will use the figure below taken from the TCGA VCF 1.1 Specification pages to summarize the components of a VCF.
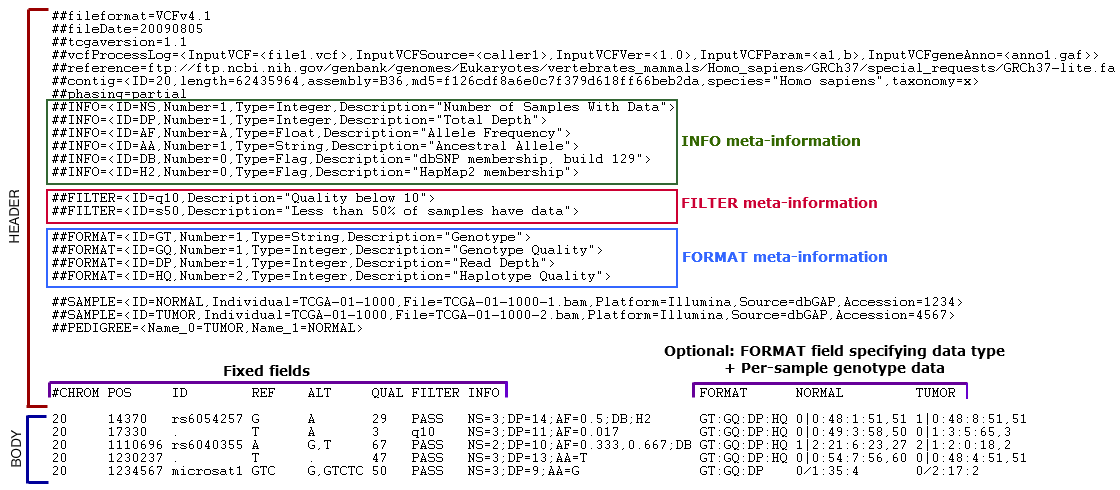
A VCF file is composed of three main parts:
1. Header
This contains meta-information lines that provide supplemental information and they always start with ##
- Lines can be applicable to all variant records in the file (e.g., date of creation of file) OR
- Lines can be specific to individual variants (e.g. flag to indicate whether a given variant exists in dbSNP)
2a. Fixed Fields
A line starting with a single # that contains headers for all columns in the Body of the file. The following eight mandatory fields will be found in every VCF file:
- CHROM - Chromosome where the variant was found
- POS - A 1-based index for the position on the chromosome where the variant was found. For multibase variants, this corresponds to the first base’s position.
- ID - If a SNP has an identifier (i.e. such as an rs number(s) from dbSNP), then it is put here. Otherwise, it will be a
.. - REF - The reference base(s) for the given position
- ALT - The variant base(s) for the given position. In the case of multiple variants present, they will be comma separated.
- QUAL - A PHRED-scaled quality score for the variant
- FILTER - A status of ‘PASS’ is given for any variant passing all filters. If a variant fails, then a semi-colon separated list will enumerate the filter(s) that the variant failed.
- INFO - Additional information about the variant. Common catergories can be found in the table below:
| Abbreviation | Data Type |
|---|---|
| AF | Allele Frequency for the ALT allele |
| DP | Combined Depth across all samples |
| NS | Number of samples with data |
2b. Optional Fields
- “FORMAT” onwards are optional fields and are included to encapsulate per-sample/genome genotype data.
- A colon-separated list of abbreviated catergories corresponding to the colon-separated genotype fields. Common catergories include:
| Abbreviation | Data Type |
|---|---|
| GT | Genotype |
| DP | Depth of the sample |
| GQ | Genotype Quality |
| HQ | Commma-separated list of Haplotype Qualities |
3. Body
These are the data lines where the variant calls will be found with each field corresponding to its column in the header line.
NOTE: For more detailed information on the VCF specification, please see our File Formats lesson where the VCF components are thoroughly described.
Exercise
First let’s move into our variant_calling directory and copy over a sample VCF file:
cd ~/variant_calling/
cp /n/groups/hbctraining/variant_calling/sample_data/sample.vcf .
1. Using grep, extract only the meta-information lines from the VCF file.
2. For the variant at position 806262 on chromosome 19, what is the reference allele?
Additional Resources
This lesson has been developed by members of the teaching team at the Harvard Chan Bioinformatics Core (HBC). These are open access materials distributed under the terms of the Creative Commons Attribution license (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.