Approximate time: 45 minutes
Learning Objectives
- Describe the ICGC-TCGA DREAM Mutation Calling Challenge Dataset
- Configure a workspace on the
/n/scratchdrive - Organize dataset for analysis
- Differentiate between using
/homeand/n/scratchdrives
Cancer genomics and the application of variant calling
Cancer is the second leading cause of death globally, so understandably there have been considerable efforts put forth to study and treat cancer. Cancer is abnormal cell growth stemming from alterations in an individual’s DNA sequence. Thus, understanding the variants observed in a given cancer can provide insight into the potential treatment options a person may undergo.
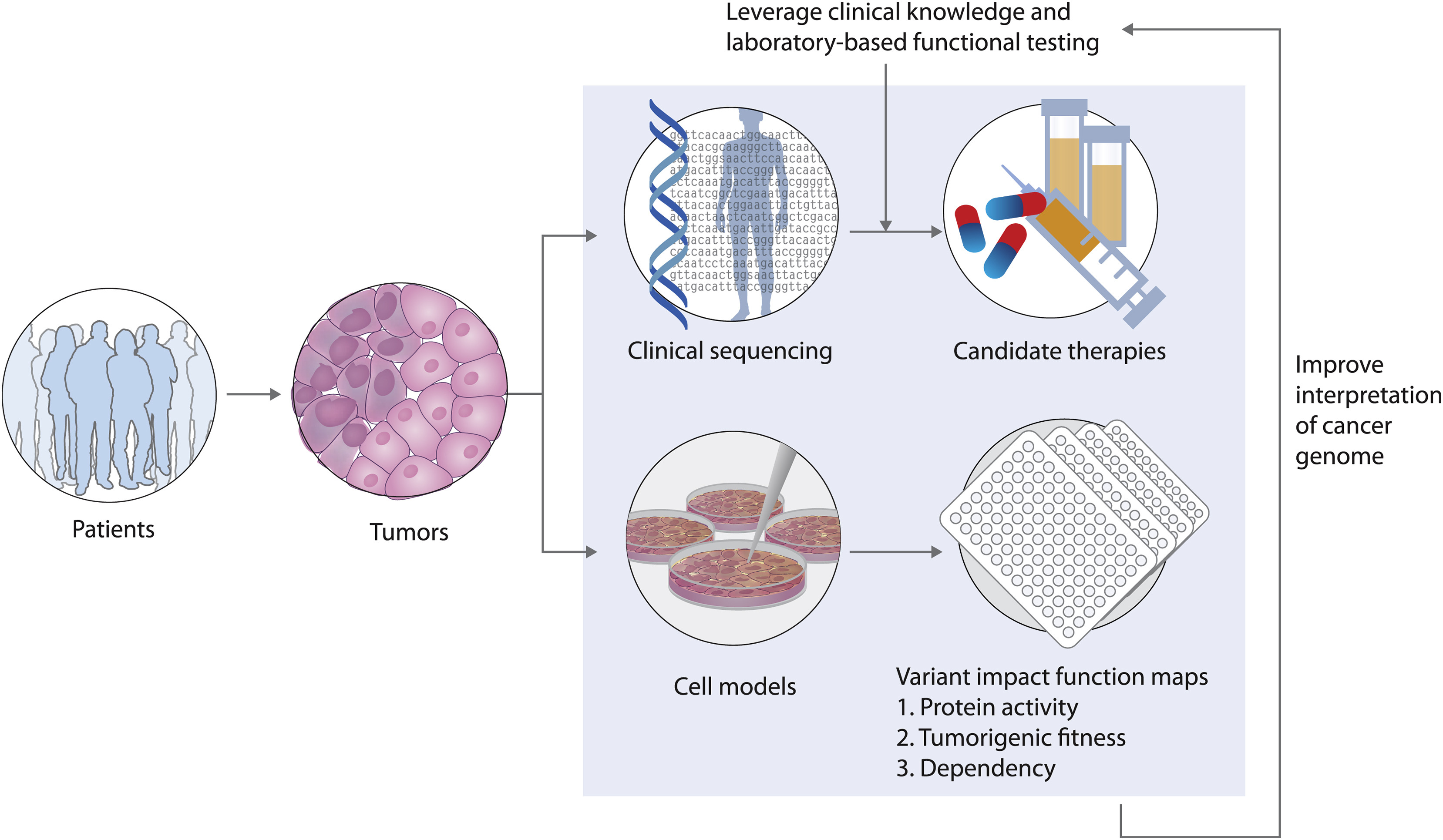
Image source: Johannessen CM and Boehm JS. Curr, Current Opinions in Systems Biology 2017
There is a vast amount of cancer genomic data deposited in public repositories which are available to researchers. These resources involve a range of large scale datasets and analysis tools, and require differing levels of computational expertise among users. Access to these resources allows us to:
- Obtain data to re-analyze and explore different questions posed from original studies
- Compare our results to large cancer databases for variant annotation
- Obtain reference datasets for benchmarking of variant calling algorithms
Genomics Data Commons
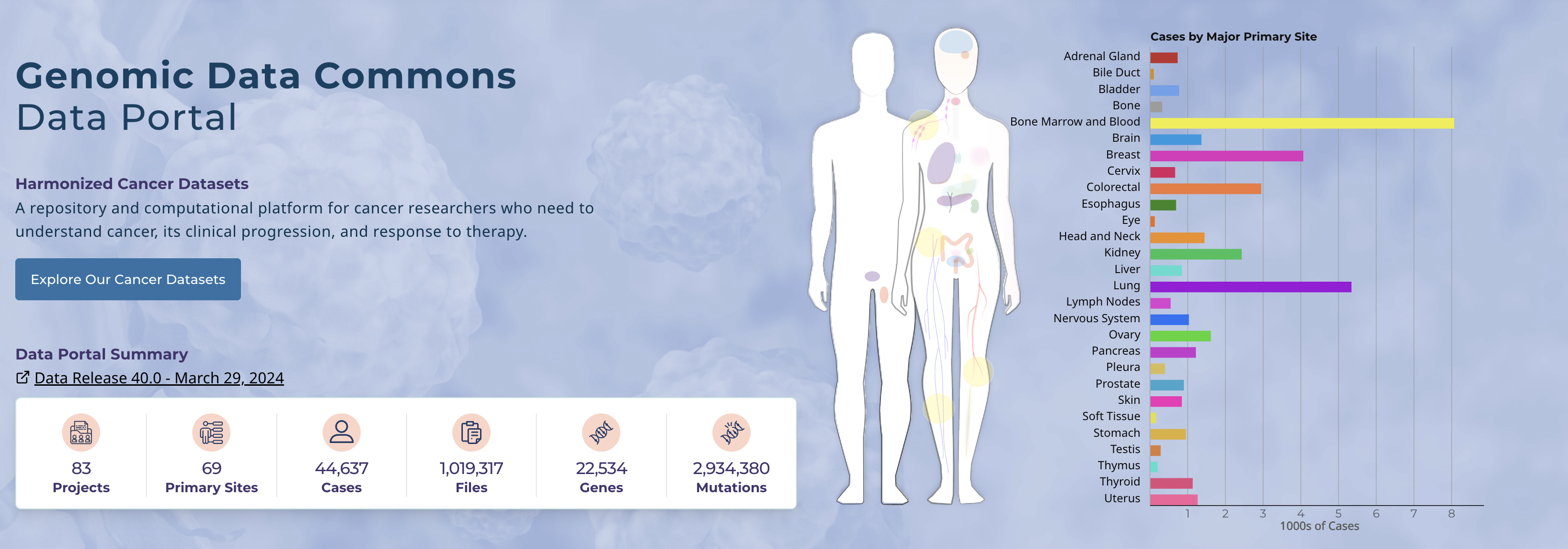
The GDC is a data repository funded by the National Cancer Institute (NCI) which provides researchers with access to genomic and clinical data from cancer patients. There is no original research conducted as part of the GDC; their main purpose is to provide centralized access to the data generated by other projects for broader research use. It contains data submitted by researchers and large scale cancer sequencing projects (such as the TCGA). The datasets go beyond whole genome sequencing, with data from RNA-seq, proteomics, imaging and other modalities. Researchers can access and query the data through the portal using built-in analysis tools, or raw data can be obtained after getting authorized access.
The Cancer Genome Atlas (TCGA)
The TCGA is a joint effort between the National Cancer Institute (NCI) and the National Human Genome Research Institute (NHGRI). The TCGA has profiled and analyzed large numbers of human tumors (across 33 different cancer types) at the DNA, RNA , protein and epigenetic levels. Researchers at the TCGA conduct the original research which includes collecting tumor samples, performing various omics analyses, and analyzing the resulting data. TCGA research has led to many discoveries about the molecular basis of cancer and identified potential biomarkers and therapeutic targets.
cBioPortal
![]()
cBioPortal is a free online resource for exploring, visualizing and analyzing cancer genomics data. Data hosted includes somatic mutations, copy number alterations, mRNA and protein expression, and clinical information for tens of thousands of cancer samples. Users can search and visualize genetic alterations across samples for specific genes or across whole cancer studies. This platform enables researchers to generate and test hypotheses, and share results with others. While there is plenty you can query via the web browser, cBioPortal also provides APIs for computational analysis and integration into other tools/pipelines. We will explore this resource in greater detail later in this workshop!
The ICGC-TCGA DREAM Mutation Calling Challenge
In 2014-2015 The Cancer Genome Atlas (TCGA) and International Cancer Genome Consortium (ICGC) co-sponsered the DREAM Mutation Calling Challenge in order to help develop methods to more accuarately predict cancer-associated mutations from whole genome resequencing data. This was a crowd-sourced competition held to evaluate algorithms for somatic mutation calling from cancer genome sequencing data in real normal/tumor datasets.
There are a few datasets that the ICGC-TCGA DREAM Mutation Calling Challenge made available as part of this challenge:
1) 10 Real Normal/Tumor datasets from anonymous cancer patients
2) 5 Synthetic Normal/Tumor datasets developed in silico
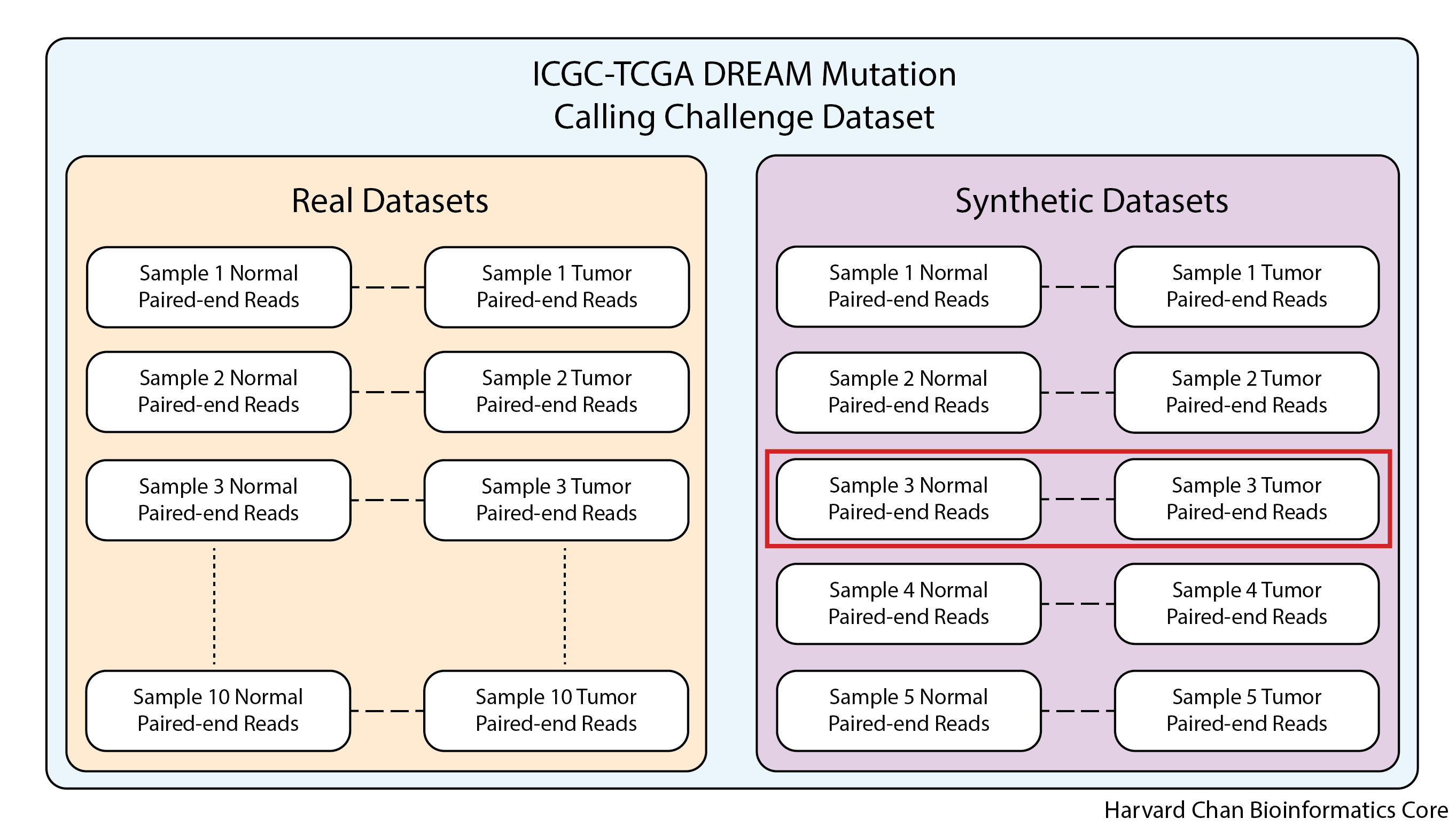
As would be expected, due to ethical standards, using the real data requires approval from ICGC and would be difficult to use in a workshop like this where we need to able to distribute the datasets to participants. Fortunately, the synthetic datasets are freely available for use and do not require ICGC approval, so this workshop will be using a single synthetic normal/tumor sample pair (synthetic dataset 3) which has multiple subclones, enabling detection of lower frequency variants.
In order to minimize resource usage in the O2 computing cluster and speed up the workflow, we will be using the whole exome sequencing (WES) dataset rather than a whole genome sequencing (WGS) dataset, but all of the methods that we will be using can be applicable to both WES and WGS datasets.
Logging into O2
For this workshop we will be using training accounts to log into O2. These have been created for us by the HMS Research Computing (HMS-RC) team and they are the folks that manage the O2 cluster. We will be providing each of you with your own training account associated with a password for the duration of this workshop. Your training account and password can be found here.
If you are interested in getting your own personal account on O2, please follow the instructions provided here after this workshop.
Let’s get started with the hands-on component by typing in the following command to log into our command-line:
ssh username@o2.hms.harvard.edu
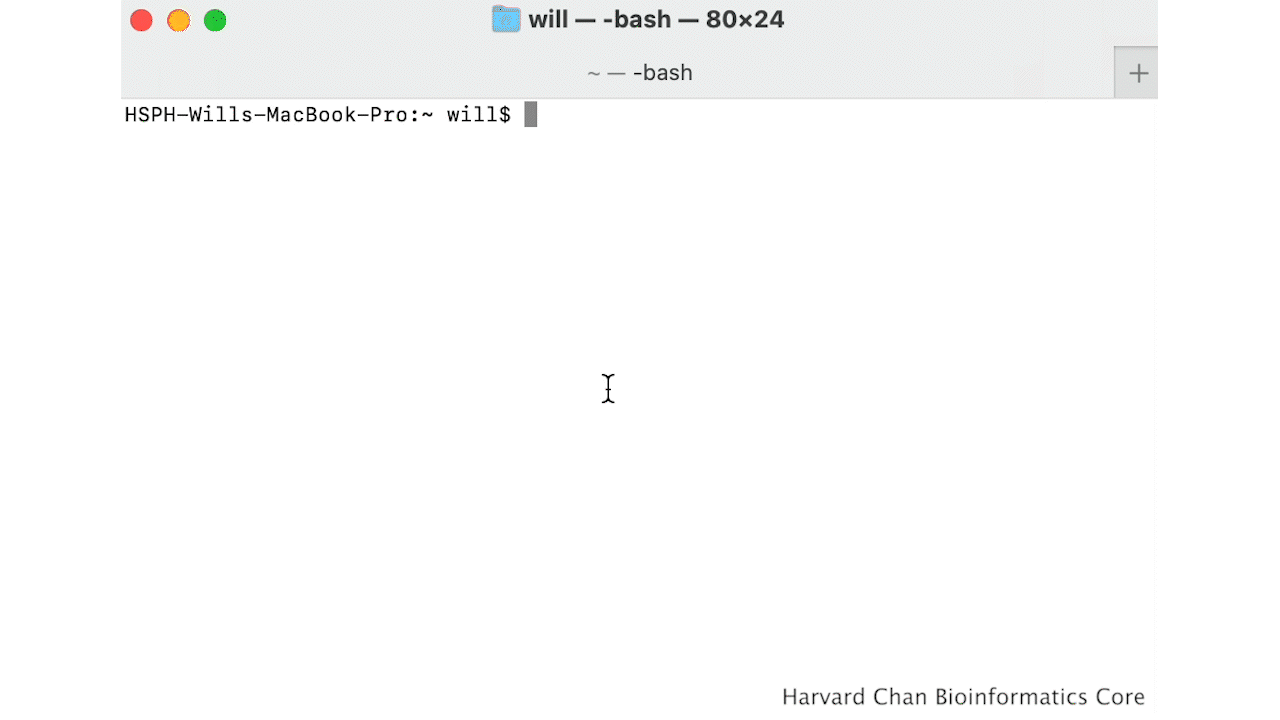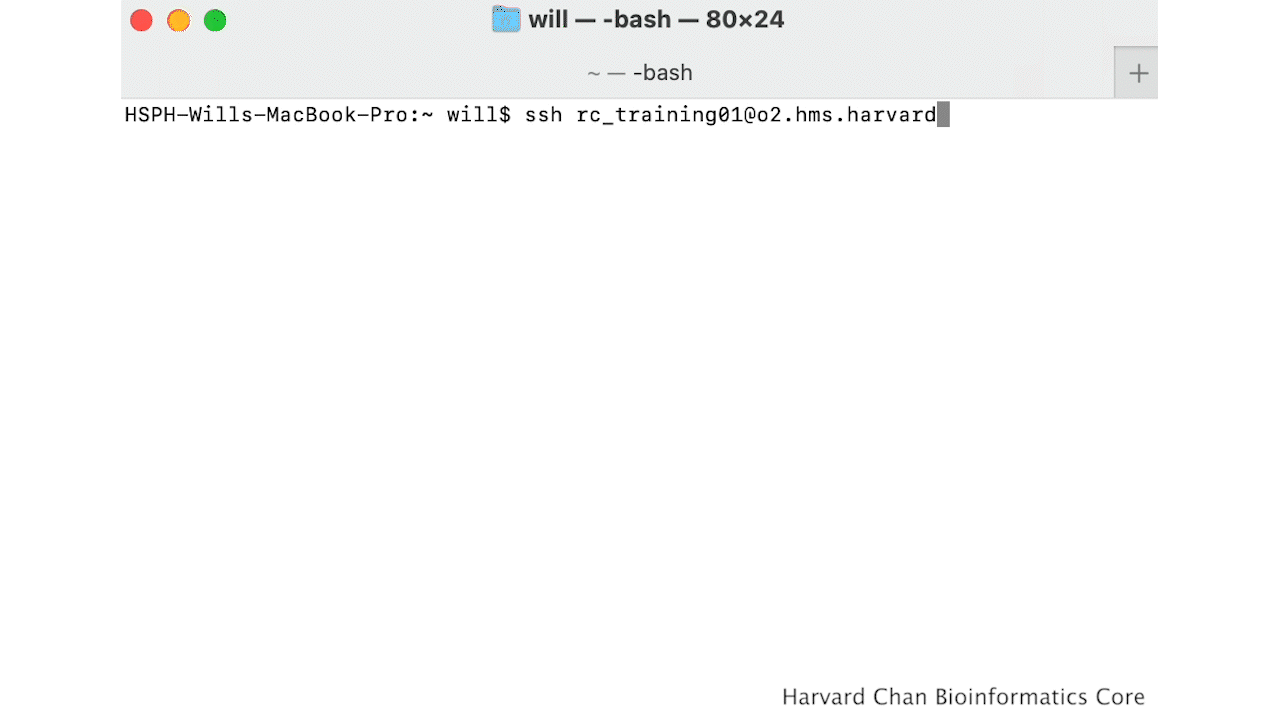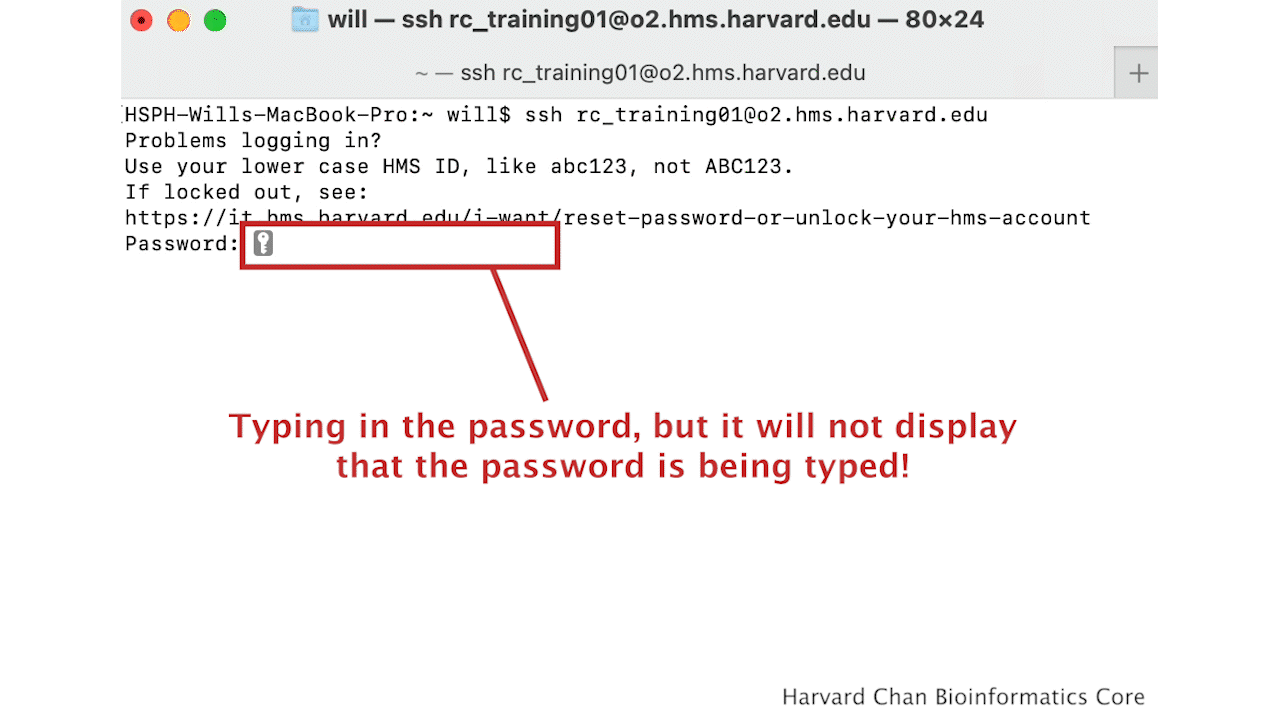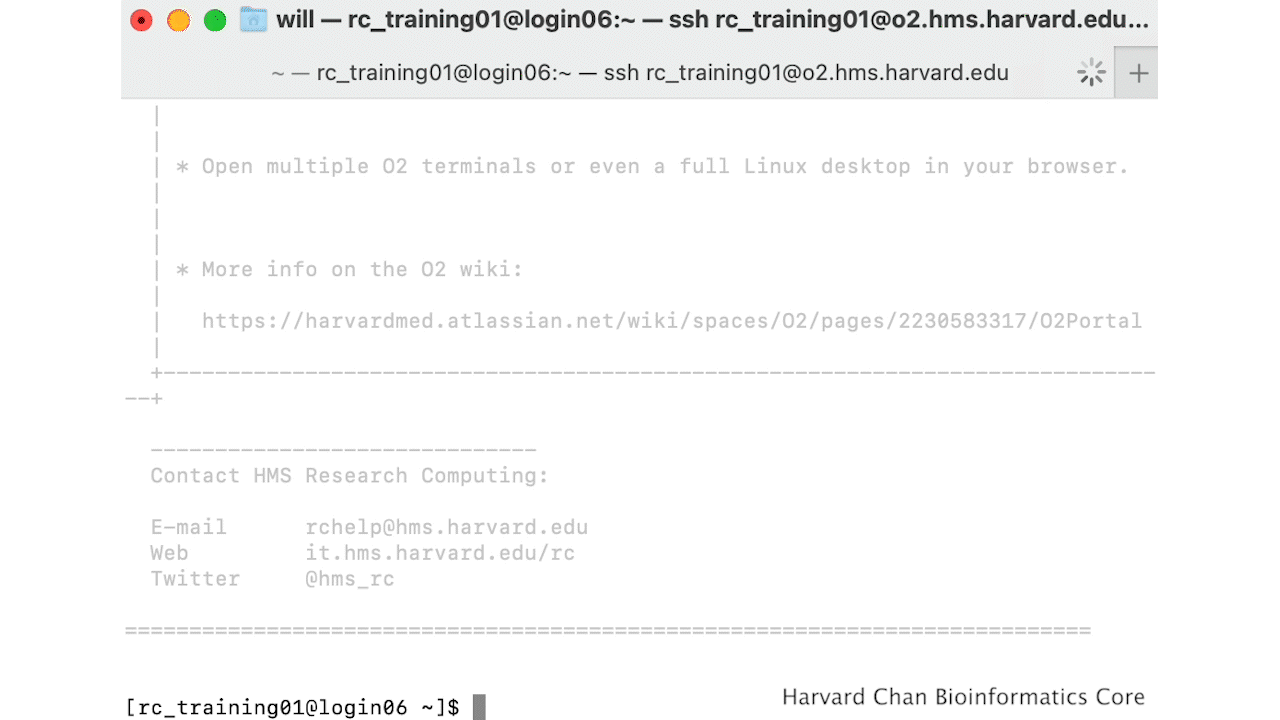
You will receive a prompt for your password, and you should type in your associated password.
Note: The cursor will not move as you type in your password.
A warning might pop up the first time you try to connect to a remote machine, type Yes or Y, then hit Enter/Return.
Once logged in, you should see the O2 icon, some news, and the command prompt, e.g. [rc_training10@login01 ~]$.
Note 1: ssh stands for secure shell. All of the information (like your password) going between your computer and the O2 login computer is encrypted when using ssh.
Creating a scratch space
Due to the limited storage space on
/home, we are going to take advantage ofscratchto hold some of our intermediate files for this workshop. This is a very common use of the scratch space as many analyses will have large intermediate files, which would otherwise fill up our home directories.
While on the login node, we will create our space on /n/scratch. In order to do so, we will need to run a script provided by the HMS Research Computing team:
$ sh /n/cluster/bin/scratch_create_directory.sh
Note: You MUST be on a login node in order to create a space on
/n/scratch.
It will prompt you with the following:
Do you want to create a scratch directory under /n/scratch/users? [y/N]>
To this you will respond y, then hit Enter/Return.
Next, it will prompt you with:
By typing 'YES' I will comply with HMS RC guidelines for using Scratch.
I also confirm that I understand that files in my scratch directory WILL NOT BE BACKED UP IN ANY WAY.
I also understand that 45 DAYS after I last modify a given file or directory in my scratch directory,
it will be DELETED with NO POSSIBILITY of retrieval.
I understand HMS RC guidelines for using scratch:
Type YES, then hit Enter/Return.
It should return:
Your scratch directory was created at /n/scratch/users/r/rc_trainingXX.
This has a limit of 25TiB of storage and 2.5 million files.
You can check your scratch quota using the quota-v2 command.
Now that we have created our scratch space, you will need to start an interactive session. A login node’s primary function is to enable users to log in to a cluster, it is not meant to be used for any actual work/computing. Since we will be doing some work, let’s get on to a compute node:
$ srun --pty -p interactive -t 0-3:00 --mem 1G /bin/bash
Make sure that your command prompt is now preceded by a character string that contains the word compute.
Implementing data management best practices
In a previous lesson, we describe the data lifecycle and the different aspects to consider when working on your own projects. Here, we implement some of those strategies to get ourselves setup before we begin with any analysis.
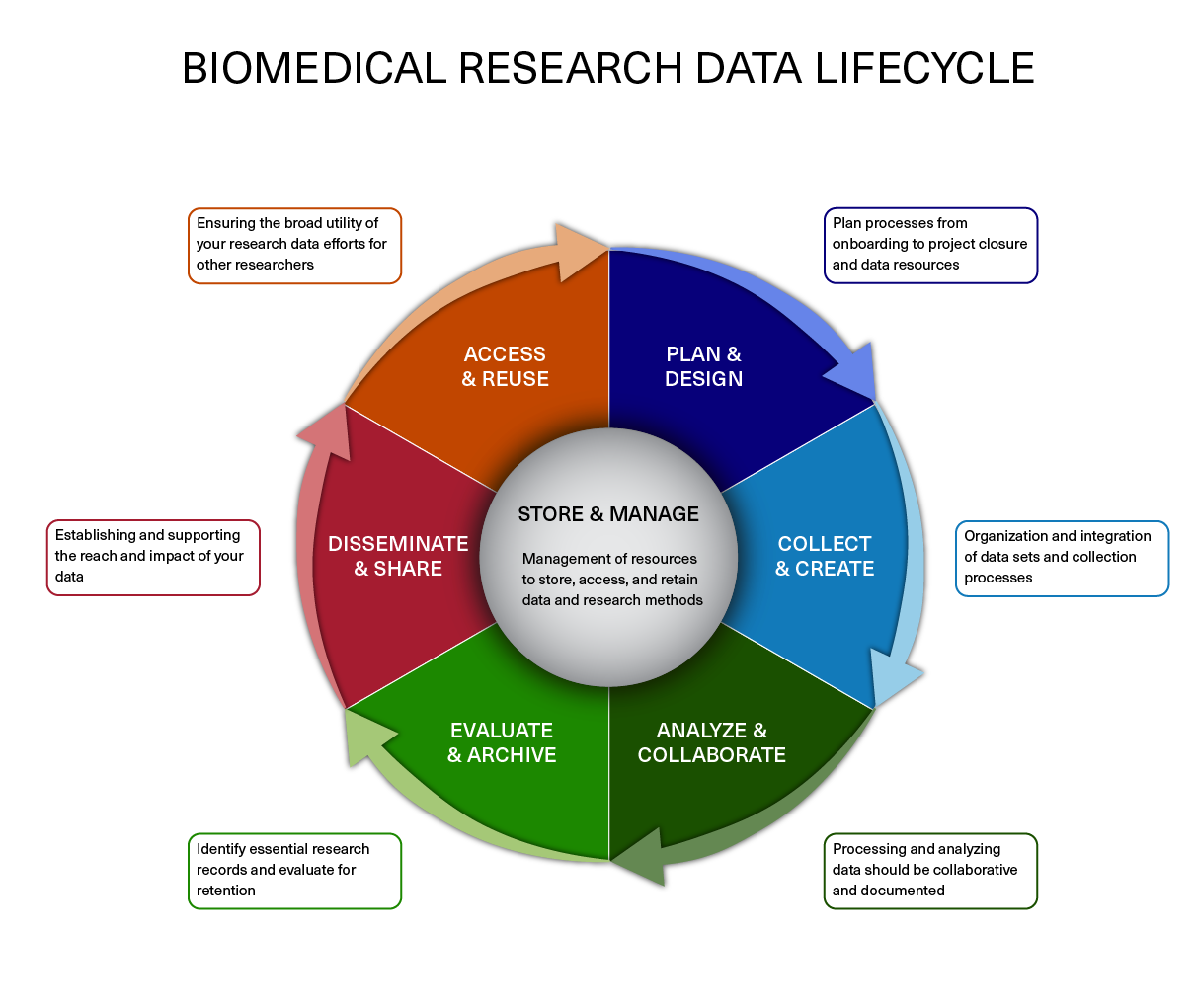
Image acquired from the Harvard Biomedical Data Management Website
Organizing our project
For each experiment you work on and analyze data for, it is considered best practice to get organized by creating a planned storage space (directory structure). We will start by creating a directory that we can use for the rest of the workshop. First, make sure that you are in your home directory.
$ cd
$ pwd
This should let us know that we are located at: /home/rc_trainingXX
Once we have established our location, let’s begin by making the directory for our project (i.e. the analysis in this workshop):
$ mkdir variant_calling
Next, we are going to move into this newly created directory and create some additional sub-directories to hold our work:
$ cd variant_calling
$ mkdir scripts results figures raw_data reports
Our project directory now has the following structure within it to keep files organized:
variant_calling/
├── figures/
├── raw_data/
├── reports/
├── results/
└── scripts/
Let’s move into our raw_data directory and copy the FASTQ files that we are going to use for our analysis:
$ cd raw_data
$ cp /n/groups/hbctraining/variant_calling/raw_data/*.fq.gz .
This may take up to a minute as there is a lot of data to copy. Now that we have created the directories that we are going to use in our home space. Let’s move to our /n/scratch space so that we can set up the directories that we are going to use to hold our intermediate files:
$ cd /n/scratch/users/${USER:0:1}/$USER
A quick explanation on the use of
${USER:0:1}The
userson the scratch space are organized into subdirectories starting with the first letter of their username.${USER:0:1}will return the first letter of the username. Test this usingecho ${USER:0:1}at the command line.This is syntax for creating substrings in
bash:${VARIABLE:START:LENGTH}
VARIABLEis the variable that you would like to create a substring ofSTARTis the 0-based indexing of the position to start the substringLENGTHis the number of characters following the starting position to include in the substring
Now that we have navigated to our scratch space, let’s go ahead and create a directory for our analysis:
$ mkdir variant_calling
Now, move inside this newly created directory and create the following directories to hold our intermediate files:
$ cd variant_calling
$ mkdir alignments vcf_files
Excellent! Our project space has now been organized and is ready for the analysis to begin!
This lesson has been developed by members of the teaching team at the Harvard Chan Bioinformatics Core (HBC). These are open access materials distributed under the terms of the Creative Commons Attribution license (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.