Self-learning Answer Key
File Formats
Using our knowledge of FLAGs in SAM files let’s decode a few using the tool on the Broad’s Website.
1. An alignment has a FLAG of 115. What do we know about this read?
Click here to see the answer
- Read paired
- Read mapped in proper pair
- Read reverse strand
- Mate reverse strand
- First in pair
2. What would be the FLAG for a read alignment for the first read in a pair-end read, where the first read was unmapped while the second read was mapped to the reverse strand?
Click here to see the answer
1013. Below is an alignment, what would be the CIGAR string for this alignment?
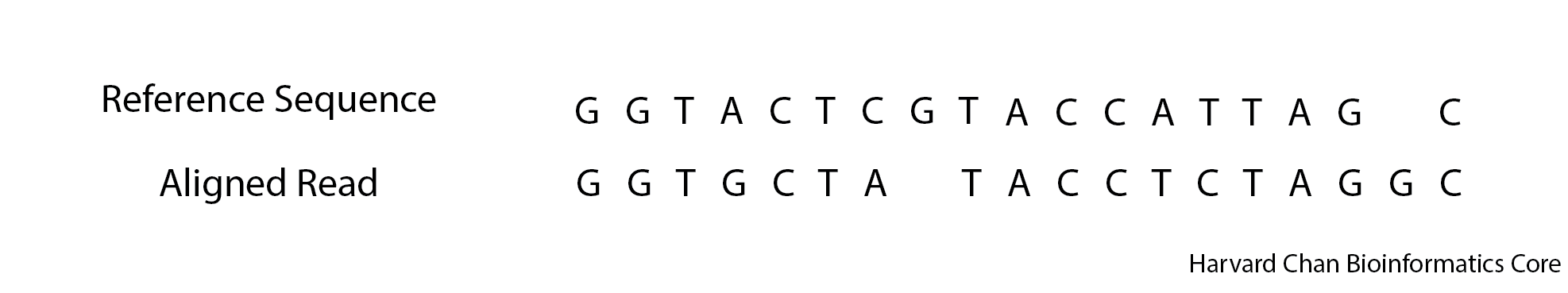
Click here to see the answer
3M1X2M1X1D4M2X2M1M1I1M4. Using grep, extract only the meta-information lines from the VCF file.
Click here to see the answer
grep '^##' sample.vcf
5. using grep, extract the lines containing the names of all of the software packages that were used in the creation of this VCF file?
Click here to see the answer
grep '^##source' sample.vcf
Bonus Challenge 6. For the sample at position 806262 on chromosome 19, what is the reference allele?
Click here to see the answer
Cless sample.vcfThen search the
less buffer with:
/19Tab806262OR
grep -e $'^19\t806262' sample.vcf
7. Create a BED file within ~/variant_calling/ called my_file.bed and have it contain these three ranges:
Chromosome 1 from 84573 to 94573
Chromosome 2 from 465352 to 466352
Chromosome 19 from 111237 to 111238
Click here to see the answer
Openvim with:
vim my_file.bedEnter insert mode and type:
1 84573 94573 2 465352 466352 19 111237 111238Exit insert mode by pressing Esc, then save and quit the file by typing
:wq while in Command mode.
Evaluating Read Qualities with FastQC
1. If the probability of a incorrect base call is 1 in 3,981, what is the associated PHRED score?
Click here to see the answer
-10 x log10(1/3981) ≈ 362. Assign the path, /The/path/to/my/vcf_file.vcf, to a variable named VCF_PATH and replace the .vcf extension with .filtered.vcf.
Click here to see the answer
VCF_PATH=/The/path/to/my/vcf_file.vcf
echo ${VCF_PATH%.vcf}.filtered.vcf
3. Assign the new path with the .filtered.vcf extension to a variable named FILTERED_VCF_PATH then echo this variable.
Click here to see the answer
FILTERED_VCF_PATH=`echo ${VCF_PATH%.vcf}.filtered.vcf`
echo $FILTERED_VCF_PATH
4. Copy the BED file from /n/groups/hbctraining/variant_calling/sample_data/sample.bed to ~/variant_calling/ directory. Move to the ~/variant_calling/ directory and use sed to stripe chr from the chromosome names and have the output look like:
1 200 300
1 600 900
2 10 1000
Click here to see the answer
cp /n/groups/hbctraining/variant_calling/sample_data/sample.bed ~/variant_calling/ cd ~/variant_calling/ sed 's/chr//g' sample.bed
5. Redirect this output to a new file called sample.without_chr.bed
Click here to see the answer
sed 's/chr//g' sample.bed > sample.without_chr.bed
Sequence Alignment Theory
1. The read group field LB (Library) is a required field to when adding read groups using Picard’s AddOrReplaceReadGroups, but we don’t currently have this field in our read group information. How would we alter out bwa command to include LB as well?
Click here to see the answer
Change:-R "@RG\tID:$SAMPLE\tPL:illumina\tPU:$SAMPLE\tSM:$SAMPLE"To:
-R "@RG\tID:$SAMPLE\tPL:illumina\tPU:$SAMPLE\tSM:$SAMPLE\tLB:$SAMPLE"
2. If we wanted to increase the number of threads used by bwa for processing our alignment to 12, where are the two places we would need to modify our SBATCH script to accommodate this?
Click here to see the answer
Change with theSBATCH directives:
#SBATCH -c 8To:
#SBATCH -c 12AND Change the
bwa command:
-t 8 \To:
-t 12 \
Alignment File Processing
1. When inspecting a SAM file you see the following order:
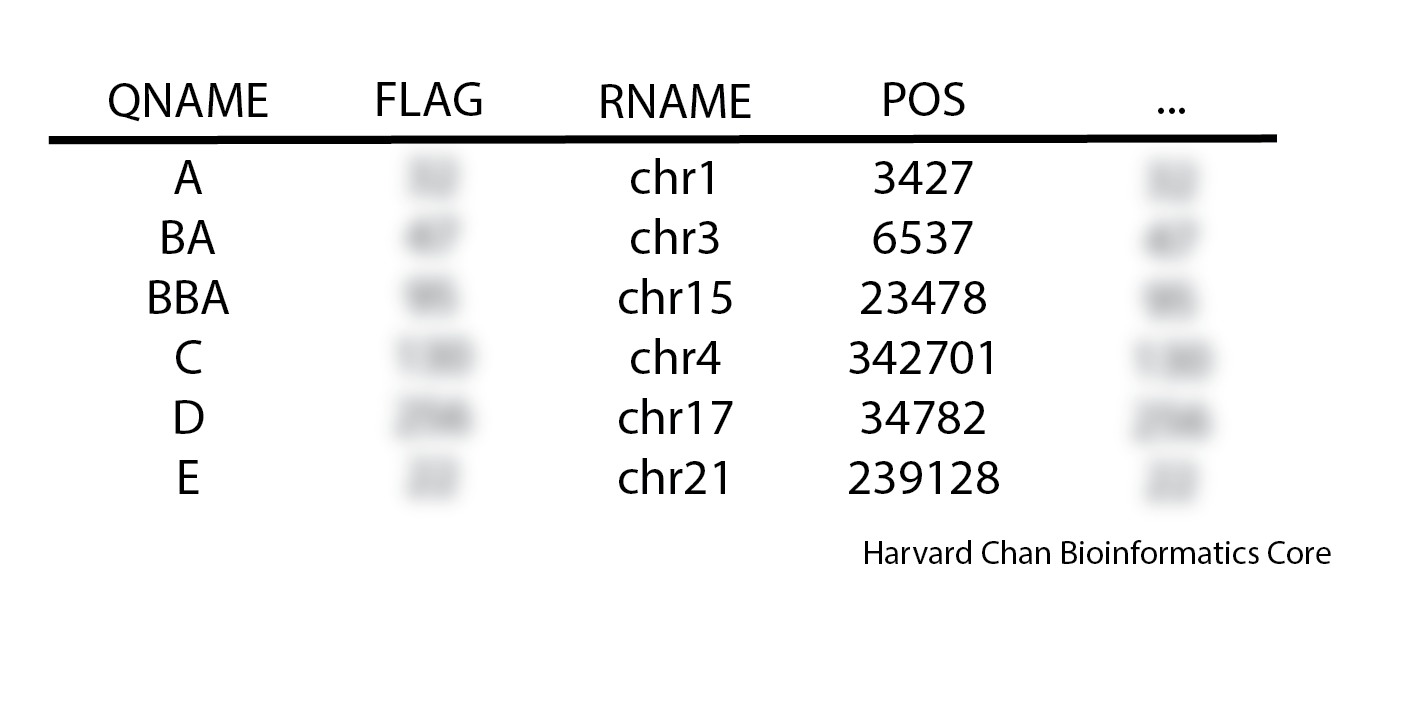
Is this SAM file’s sort order: unsorted, query-sorted, coordinate-sorted or is it ambiguous?
Click here to see the answer
Query-sorted2. We are comparing our SortSam command with our colleague’s command. Is there anything wrong with their syntax? Why or why not?
Our syntax
java -jar $PICARD/picard.jar SortSam \
--INPUT $SAM_FILE \
--OUTPUT $QUERY_SORTED_BAM_FILE \
--SORT_ORDER queryname
Our colleague’s syntax
java -jar $PICARD/picard.jar SortSam \
I=$SAM_FILE \
O=$QUERY_SORTED_BAM_FILE \
SO=queryname
Click here to see the answer
No, it is just using the traditional syntax with abbreviations.Alignment Quality Control
1. Inspect your coordinate-sorted BAM file with ViewSam package within Picard. What version of bwa was used in the alignment?
Click here to see the answer
bwa version 0.7.17-r11882. Inspect your coordinate-sorted BAM file with ViewSam package within Picard. What is the flag for your first aligned read? Using the Broad’s decoding FLAG tool, what does this flag mean?
Click here to see the answer
129- Read paired
- Second in pair
Automation of Variant Calling
1. If we submit Job_A.sh and SLURM returns:
Submitted batch job 213489
And we want to start Job_B.sh after Job_A.sh finishes without error, what command would we use to do this?
Click here to see the answer
sbatch --dependency=afterok:213489 Job_B.sh
2. If we submit Job_X.sh and SLURM returns:
Submitted batch job 213489
Then we submit Job_Y.sh and SLURM returns:
Submitted batch job 213496
And we want to start Job_Z.sh after Job_X.sh and Job_Y.sh finishes without error, what command would we use to do this?
Click here to see the answer
sbatch --dependency=afterok:213489:213496 Job_Z.sh
3. If you want to submit Job_M.sh, and provide reference.fa as the first positional parameter and input.bam as the second positional parameter, how could you do this?
Click here to see the answer
sbatch Job_M.sh reference.fa input.bam
Variant Prioritization
1. Extract from mutect2_syn3_normal_syn3_tumor_GRCh38.p7-pass-filt-LCR.pedigree_header.snpeff.dbSNP.vcf all of the MODERATE-impact mutations on Chromosome 12.
Click here to see the answer
java -jar $SNPEFF/SnpSift.jar filter "( CHROM = '12' ) & ( ANN[*].IMPACT has 'MODERATE' )" mutect2_syn3_normal_syn3_tumor_GRCh38.p7-pass-filt-LCR.pedigree_header.snpeff.dbSNP.vcf | less
2. Pipe the output from Exercise 1) into a command to only display one line for each effect.
Click here to see the answer
java -jar $SNPEFF/SnpSift.jar filter "( CHROM = '12' ) & ( ANN[*].IMPACT has 'MODERATE' )" mutect2_syn3_normal_syn3_tumor_GRCh38.p7-pass-filt-LCR.pedigree_header.snpeff.dbSNP.vcf | $SNPEFF/scripts/vcfEffOnePerLine.pl | less
3. Pipe the output from Exercise 2) into a command to extract the chromosome, position, gene and effect.
Click here to see the answer
java -jar $SNPEFF/SnpSift.jar filter "( CHROM = '12' ) & ( ANN[*].IMPACT has 'MODERATE' )" mutect2_syn3_normal_syn3_tumor_GRCh38.p7-pass-filt-LCR.pedigree_header.snpeff.dbSNP.vcf | $SNPEFF/scripts/vcfEffOnePerLine.pl | java -jar $SNPEFF/SnpSift.jar extractFields - "CHROM" "POS" "ANN[*].GENE" "ANN[*].EFFECT" | less
4. Redirect the output from Exercise 3) into a new file called mutect2_syn3_normal_syn3_tumor_GRCh38.p7-pass-filt-LCR.pedigree_header.snpeff.dbSNP.chr12_moderate-impact.txt
Click here to see the answer
java -jar $SNPEFF/SnpSift.jar filter "( CHROM = '12' ) & ( ANN[*].IMPACT has 'MODERATE' )" mutect2_syn3_normal_syn3_tumor_GRCh38.p7-pass-filt-LCR.pedigree_header.snpeff.dbSNP.vcf | $SNPEFF/scripts/vcfEffOnePerLine.pl | java -jar $SNPEFF/SnpSift.jar extractFields - "CHROM" "POS" "ANN[*].GENE" "ANN[*].EFFECT" > mutect2_syn3_normal_syn3_tumor_GRCh38.p7-pass-filt-LCR.pedigree_header.snpeff.dbSNP.chr12_moderate-impact.txt