Approximate time: 30 minutes
Single-cell RNA-seq: Integration with Harmony
Overview of Harmony
In this section, we illustrate the use of Harmony as a possible alternative to the Seurat integration workflow. Compared to other algorithms, Harmony notably presents the following advantages (Korsunsky et al. 2019, Tran et al. 2020):
- Possibility to integrate data across several variables (for example, by experimental batch and by condition)
- Significant gain in speed and lower memory requirements for integration of large datasets
- Interoperability with the
Seuratworkflow
Instead of using CCA, Harmony applies a transformation to the principal component (PCs) values, using all available PCs, e.g. as pre-computed within the Seurat workflow. In this space of transformed PCs, Harmony uses k-means clustering to delineate clusters, seeking to define clusters with maximum “diversity”. The diversity of each cluster reflects whether it contains balanced amounts of cells from each of the batches (donor, condition, tissue, technolgy…) we seek to integrate on, as should be observed in a well-integrated dataset. After defining diverse clusters, Harmony determines how much a cell’s batch identity impacts on its PC coordinates, and applies a correction to “shift” the cell towards the centroid of the cluster it belongs to. Cells are projected again using these corrected PCs, and the process is repeated iteratively until convergence.
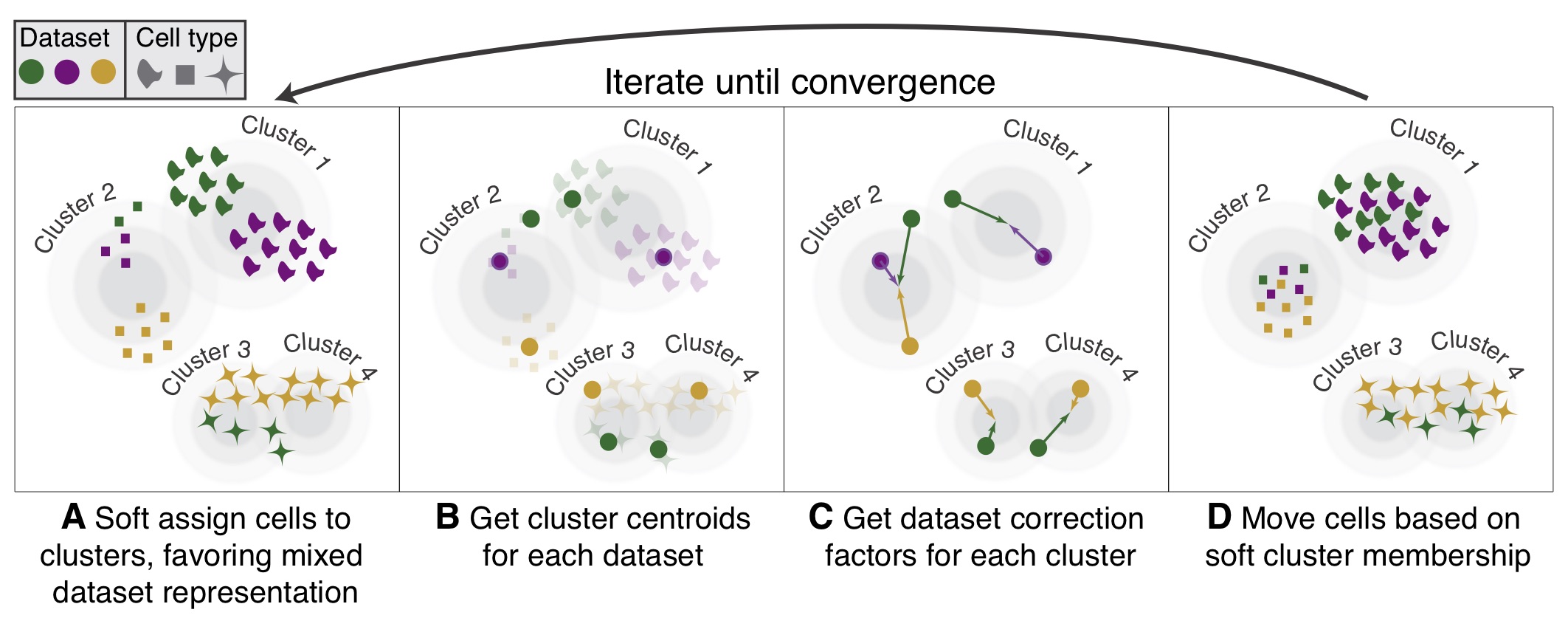
Image credit: Korsunsky, I., Millard, N., Fan, J. et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296 (2019). https://doi.org/10.1038/s41592-019-0619-0
For a more detailed breakdown of the Harmony algorithm, we recommend checking this advanced vignette from the package developers.
Implementing Harmony within the Seurat workflow
In practice, we can easily use Harmony within our Seurat workflow. To perform integration, Harmony takes as input a merged Seurat object, containing data that has been appropriately normalized (i.e. here, normalized using SCTransform) and for which highly variable features and PCs are defined.
There are 2 ways to reach that point:
- Merge the raw Seurat objects for all samples to integrate; then perform normalization, variable feature selection and PC calculation on this merged object (workflow recommended by
Harmonydevelopers) - Perform (SCT) normalization independently on each sample and find integration features across samples using
Seurat; then merge these normalized Seurat objects, set variable features manually to integration features, and finally calculate PCs on this merged object (workflow best reflecting recommendations for application ofSCTransform)
In the first scenario, assuming raw_seurat_list is a list of N samples containing raw data that have only undergone QC filtering, we would thus run the following code:
# Merge raw samples
merged_seurat <- merge(x = raw_seurat_list[[1]],
y = raw_seurat_list[2:length(raw_seurat_list)],
merge.data = TRUE)
# Perform log-normalization and feature selection, as well as SCT normalization on global object
merged_seurat <- merged_seurat %>%
NormalizeData() %>%
FindVariableFeatures(selection.method = "vst", nfeatures = 2000) %>%
ScaleData() %>%
SCTransform(vars.to.regress = c("mitoRatio"))
# Calculate PCs using variable features determined by SCTransform (3000 by default)
merged_seurat <- RunPCA(merged_seurat, assay = "SCT", npcs = 50)
In the second scenario, assuming norm_seurat_list is a list of N samples similar to our split_seurat object, i.e. containing data that have been normalized as demonstrated in the previous lecture on SCT normalization, we would thus run the following code:
# Find most variable features across samples to integrate
integ_features <- SelectIntegrationFeatures(object.list = norm_seurat_list, nfeatures = 3000)
# Merge normalized samples
merged_seurat <- merge(x = norm_seurat_list[[1]],
y = norm_seurat_list[2:length(raw_seurat_list)],
merge.data = TRUE)
DefaultAssay(merged_seurat) <- "SCT"
# Manually set variable features of merged Seurat object
VariableFeatures(merged_seurat) <- integ_features
# Calculate PCs using manually set variable features
merged_seurat <- RunPCA(merged_seurat, assay = "SCT", npcs = 50)
NOTE: As mentioned above, there is active discussion within the community regarding which of those 2 approaches to use (see for example here and here). We recommend that you check GitHub forums to make your own opinion and for updates.
Regardless of the approach, we now have a merged Seurat object containing normalized data for all the samples we need to integrate, as well as defined variable features and PCs.
One last thing we need to do before running Harmony is to make sure that the metadata of our Seurat object contains one (or several) variable(s) describing the factor(s) we want to integrate on (e.g. one variable for sample_id, one variable for experiment_date).
We’re then ready to run Harmony!
harmonized_seurat <- RunHarmony(merged_seurat,
group.by.vars = c("sample_id", "experiment_date"),
reduction = "pca", assay.use = "SCT", reduction.save = "harmony")
NOTE: You can specify however many variables to integrate on using the
group.by.varsparameter, although we would recommend keeping these to the minimum necessary for your study.
The line of code above adds a new reduction of 50 “harmony components” (~ corrected PCs) to our Seurat object, stored in harmonized_seurat@reductions$harmony.
To make sure our Harmony integration is reflected in the data visualization, we still need to generate a UMAP derived from these harmony embeddings instead of PCs:
harmonized_seurat <- RunUMAP(harmonized_seurat, reduction = "harmony", assay = "SCT", dims = 1:40)
Finally, when running the clustering analysis later on (see next lecture for details), we will also need to set the reduction to use as “harmony” (instead of “pca” by default).
harmonized_seurat <- FindNeighbors(object = harmonized_seurat, reduction = "harmony")
harmonized_seurat <- FindClusters(harmonized_seurat, resolution = c(0.2, 0.4, 0.6, 0.8, 1.0, 1.2))
The rest of the Seurat workflow and downstream analyses after integration using Harmony can then proceed without further amendments.
This lesson has been developed by members of the teaching team at the Harvard Chan Bioinformatics Core (HBC). These are open access materials distributed under the terms of the Creative Commons Attribution license (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
- A portion of these materials and hands-on activities were adapted from the Satija Lab’s Seurat - Guided Clustering Tutorial