Approximate time: 90 minutes
Learning Objectives:
- Perform integration of cells across conditions to identify cells that are similar to each other
- Describe complex integration tasks and alternative tools for integration
Single-cell RNA-seq clustering analysis: Integration
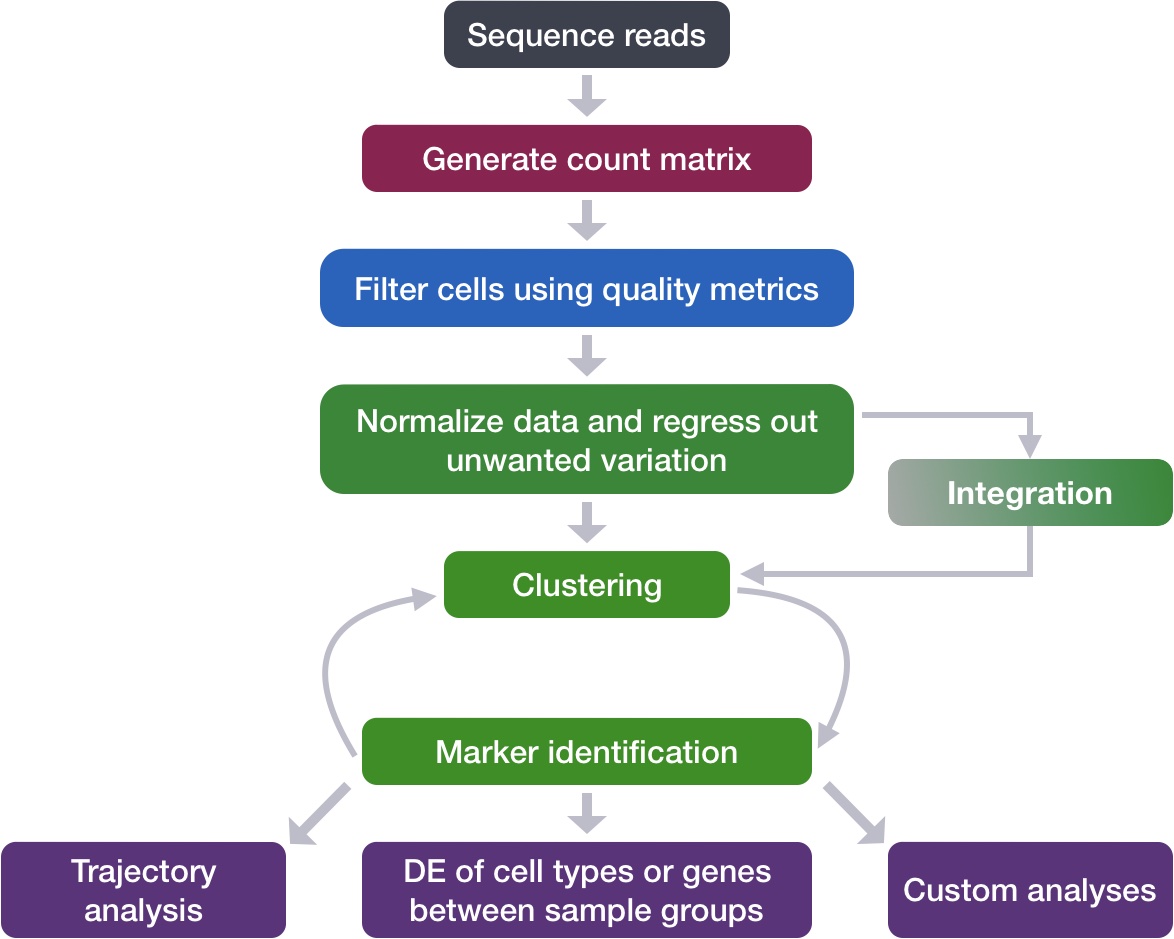
Goals:
- To align same cell types across conditions.
Challenges:
- Aligning cells of similar cell types so that we do not have clustering downstream due to differences between samples, conditions, modalities, or batches
Recommendations:
- Go through the analysis without integration first to determine whether integration is necessary
To integrate or not to integrate?
Generally, we always look at our clustering without integration before deciding whether we need to perform any alignment. Do not just always perform integration because you think there might be differences - explore the data. If we had performed the normalization on both conditions together in a Seurat object and visualized the similarity between cells, we would have seen condition-specific clustering:
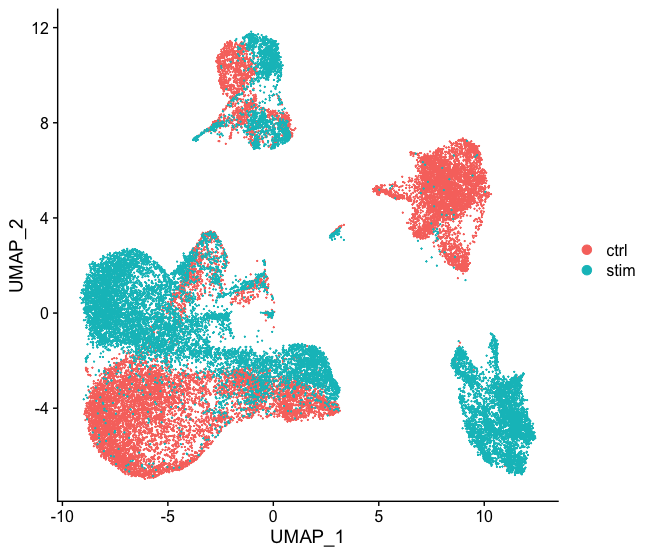
How do we create this UMAP?
The UMAP above can be generated by using the `seurat_phase` object from the previous lesson. For this object we have already run PCA, so the next steps would be to `RunUMAP()` and then plot, as shown in the code below.
# Run UMAP
seurat_phase <- RunUMAP(seurat_phase,
dims = 1:40,reduction = "pca")
# Plot UMAP
DimPlot(seurat_phase)
Condition-specific clustering of the cells indicates that we need to integrate the cells across conditions to ensure that cells of the same cell type cluster together.
Why is it important the cells of the same cell type cluster together?
We want to identify cell types which are present in all samples/conditions/modalities within our dataset, and therefore would like to observe a representation of cells from both samples/conditions/modalities in every cluster. This will enable more interpretable results downstream (i.e. DE analysis, ligand-receptor analysis, differential abundance analysis…).
In this lesson, we will cover the integration of our samples across conditions, which is adapted from the Seurat Guided Integration Tutorial.
NOTE: Seurat has a vignette for how to run through the workflow from normalization to clustering without integration. Other steps in the workflow remain fairly similar, but the samples would not necessarily be split in the beginning and integration would not be performed.
It can help to first run conditions individually if unsure what clusters to expect or expecting some different cell types between conditions (e.g. tumor and control samples), then run them together to see whether there are condition-specific clusters for cell types present in both conditions. Oftentimes, when clustering cells from multiple conditions there are condition-specific clusters and integration can help ensure the same cell types cluster together.
Integrate or align samples across conditions using shared highly variable genes
If cells cluster by sample, condition, batch, dataset, modality, this integration step can greatly improve the clustering and the downstream analyses.
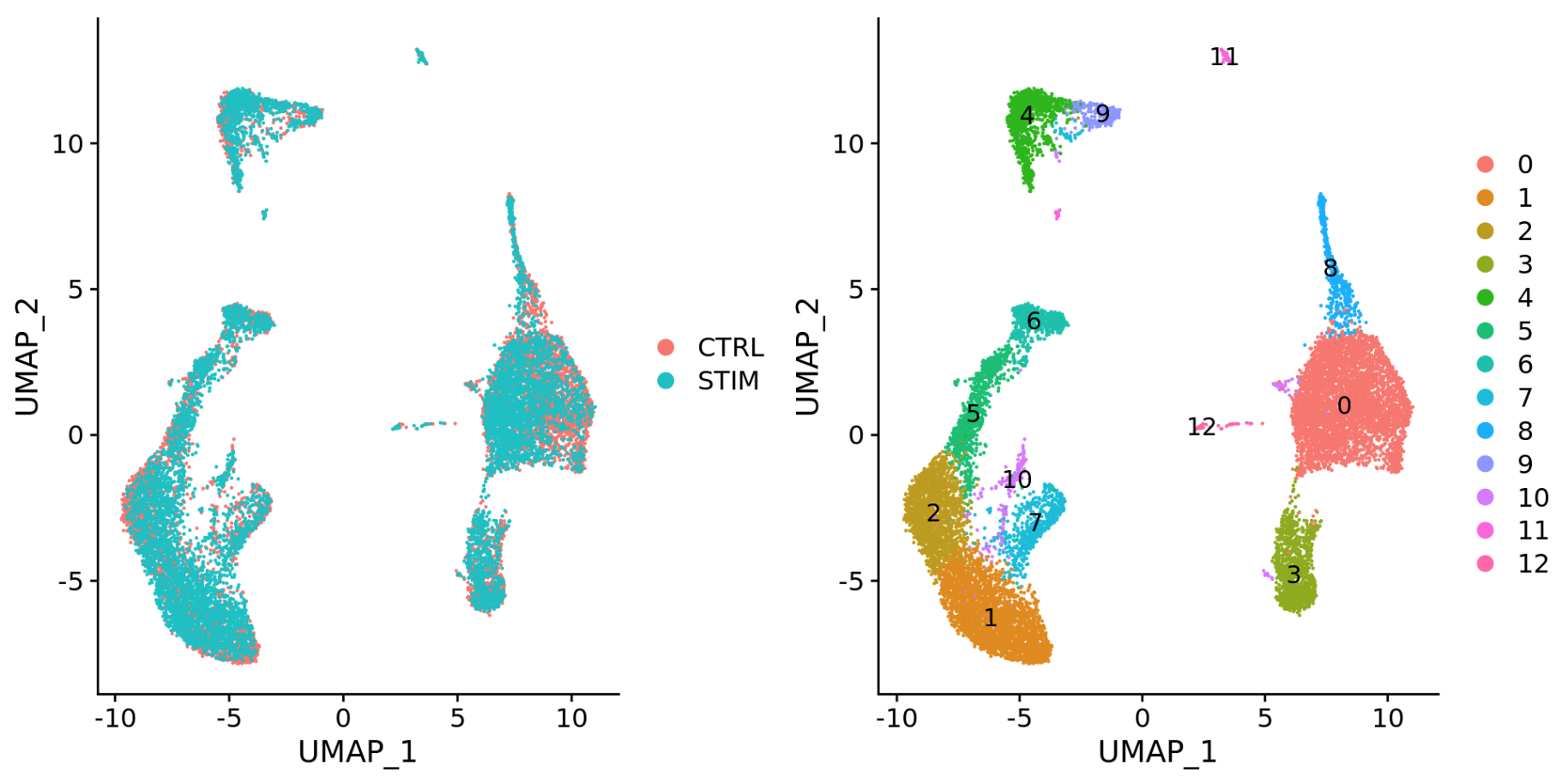
To integrate, we will use the shared highly variable genes (identified using SCTransform) from each group, then, we will “integrate” or “harmonize” the groups to overlay cells that are similar or have a “common set of biological features” between groups. For example, we could integrate across:
-
Different conditions (e.g. control and stimulated)
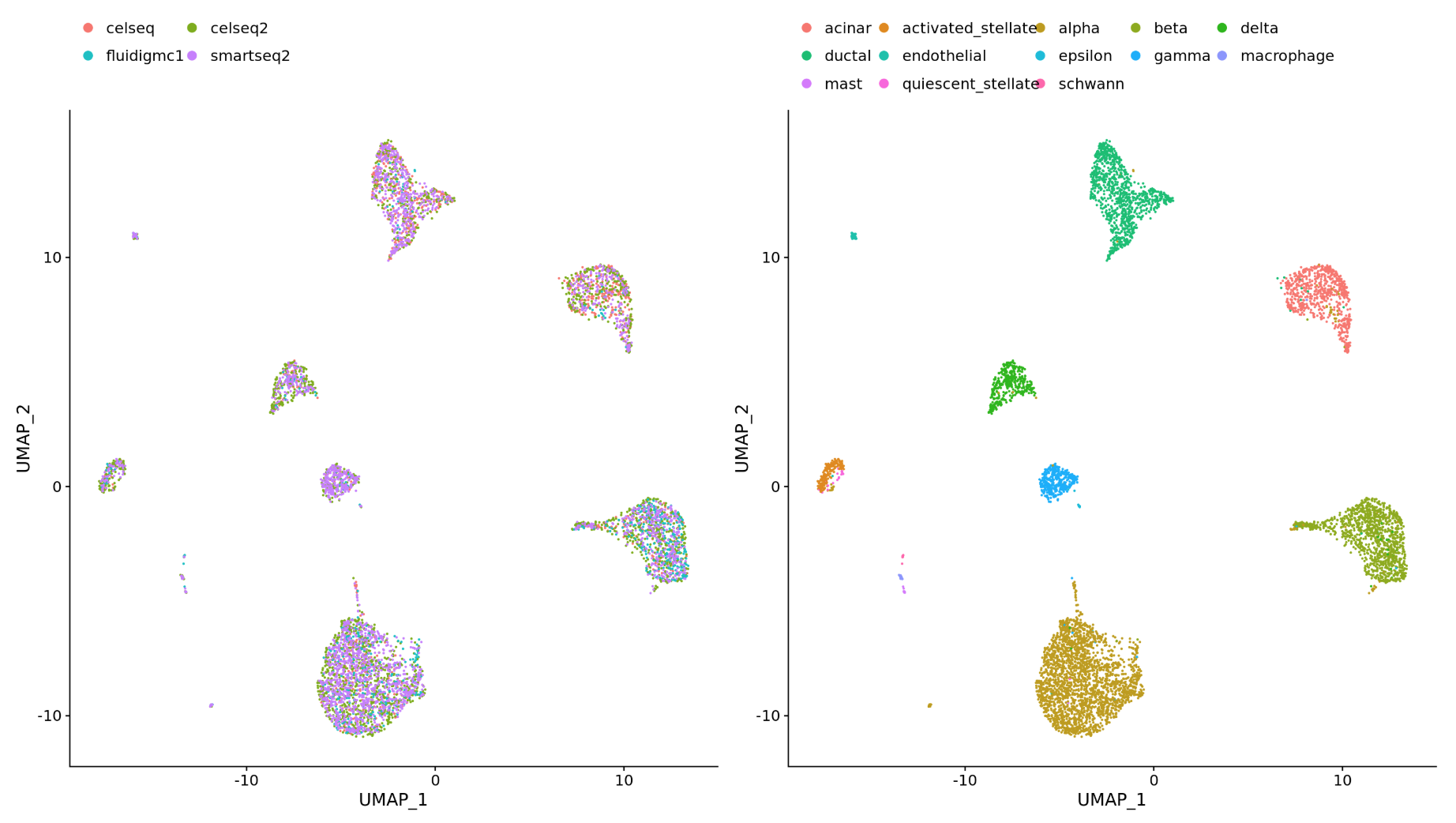
-
Different datasets (e.g. scRNA-seq from datasets generated using different library preparation methods on the same samples)
-
Different modalities (e.g. scRNA-seq and scATAC-seq)
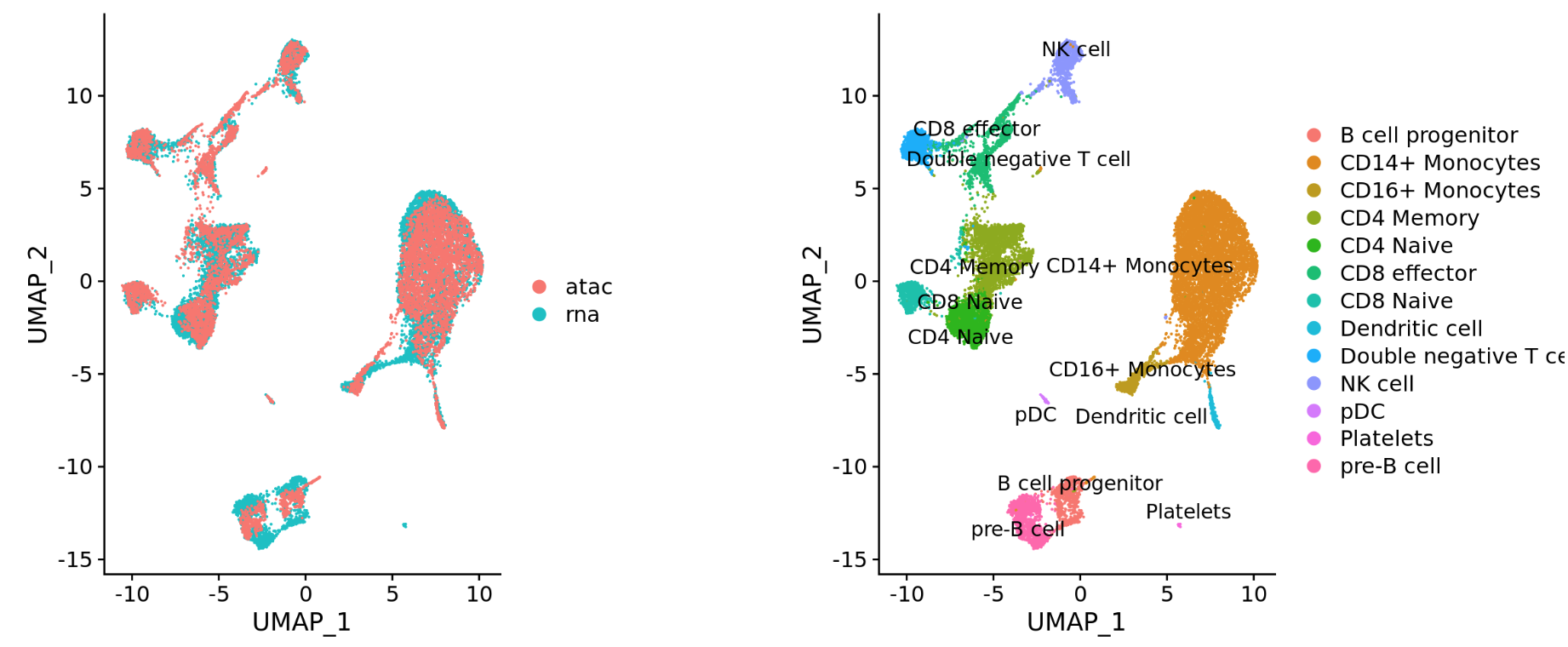
-
Different batches (e.g. when experimental conditions make batch processing of samples necessary)
Integration is a powerful method that uses these shared sources of greatest variation to identify shared subpopulations across conditions or datasets [Stuart and Bulter et al. (2018)]. The goal of integration is to ensure that the cell types of one condition/dataset align with the same celltypes of the other conditions/datasets (e.g. control macrophages align with stimulated macrophages).
Integration using CCA
The integration method that is available in the Seurat package utilizes the canonical correlation analysis (CCA). This method expects “correspondences” or shared biological states among at least a subset of single cells across the groups. The steps in the Seurat integration workflow are outlined in the figure below:
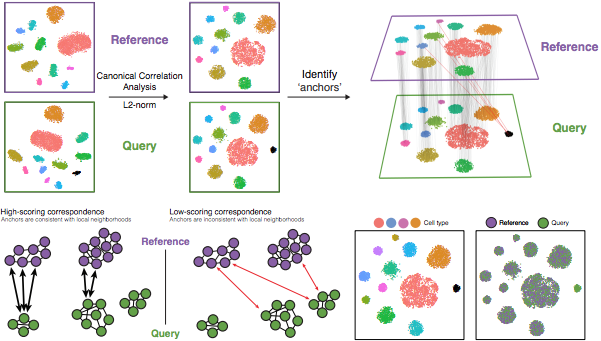
Image credit: Stuart T and Butler A, et al. Comprehensive integration of single cell data, bioRxiv 2018 (https://doi.org/10.1101/460147)
The different steps applied are as follows:
-
Perform canonical correlation analysis (CCA):
CCA identifies shared sources of variation between the conditions/groups. It is a form of PCA, in that it identifies the greatest sources of variation in the data, but only if it is shared or conserved across the conditions/groups (using the 3000 most variant genes from each sample).
This step roughly aligns the cells using the greatest shared sources of variation.
NOTE: The shared highly variable genes are used because they are the most likely to represent those genes distinguishing the different cell types present.
-
Identify anchors or mutual nearest neighbors (MNNs) across datasets (sometimes incorrect anchors are identified):
MNNs can be thought of as ‘best buddies’. For each cell in one condition:
- The cell’s closest neighbor in the other condition is identified based on gene expression values - its ‘best buddy’.
- The reciprocal analysis is performed, and if the two cells are ‘best buddies’ in both directions, then those cells will be marked as anchors to ‘anchor’ the two datasets together.
“The difference in expression values between cells in an MNN pair provides an estimate of the batch effect, which is made more precise by averaging across many such pairs. A correction vector is obtained and applied to the expression values to perform batch correction.” [Stuart and Bulter et al. (2018)].
-
Filter anchors to remove incorrect anchors:
Assess the similarity between anchor pairs by the overlap in their local neighborhoods (incorrect anchors will have low scores) - do the adjacent cells have ‘best buddies’ that are adjacent to each other?
-
Integrate the conditions/datasets:
Use anchors and corresponding scores to transform the cell expression values, allowing for the integration of the conditions/datasets (different samples, conditions, datasets, modalities)
NOTE: Transformation of each cell uses a weighted average of the two cells of each anchor across anchors of the datasets. Weights determined by cell similarity score (distance between cell and k nearest anchors) and anchor scores, so cells in the same neighborhood should have similar correction values.
If cell types are present in one dataset, but not the other, then the cells will still appear as a separate sample-specific cluster.
Now, using our SCTransform object as input, let’s perform the integration across conditions.
First, we need to specify that we want to use all of the 3000 most variable genes identified by SCTransform for the integration. By default, this function only selects the top 2000 genes.
## Don't run this during class
# Select the most variable features to use for integration
integ_features <- SelectIntegrationFeatures(object.list = split_seurat,
nfeatures = 3000)
NOTE: If you are missing the
split_seuratobject, you can load it from yourdatafolder:# Load the split seurat object into the environment split_seurat <- readRDS("data/split_seurat.rds")If you do not have the
split_seurat.rdsfile in yourdatafolder, you can right-click here to download it to thedatafolder (it may take a bit of time to download).
Now, we need to prepare the SCTransform object for integration.
## Don't run this during class
# Prepare the SCT list object for integration
split_seurat <- PrepSCTIntegration(object.list = split_seurat,
anchor.features = integ_features)
Now, we are going to perform CCA, find the best buddies or anchors and filter incorrect anchors. For our dataset, this will take up to 15 minutes to run. Also, note that the progress bar in your console will stay at 0%, but know that it is actually running.
## Don't run this during class
# Find best buddies - can take a while to run
integ_anchors <- FindIntegrationAnchors(object.list = split_seurat,
normalization.method = "SCT",
anchor.features = integ_features)
Finally, we can integrate across conditions.
## Don't run this during class
# Integrate across conditions
seurat_integrated <- IntegrateData(anchorset = integ_anchors,
normalization.method = "SCT")
UMAP visualization
After integration, to visualize the integrated data we can use dimensionality reduction techniques, such as PCA and Uniform Manifold Approximation and Projection (UMAP). While PCA will determine all PCs, we can only plot two at a time. In contrast, UMAP will take the information from any number of top PCs to arrange the cells in this multidimensional space. It will take those distances in multidimensional space and plot them in two dimensions working to preserve local and global structure. In this way, the distances between cells represent similarity in expression. If you wish to explore UMAP in more detail, this post is a nice introduction to UMAP theory.
To generate these visualizations we need to first run PCA and UMAP methods. Let’s start with PCA.
# Run PCA
seurat_integrated <- RunPCA(object = seurat_integrated)
# Plot PCA
PCAPlot(seurat_integrated,
split.by = "sample")
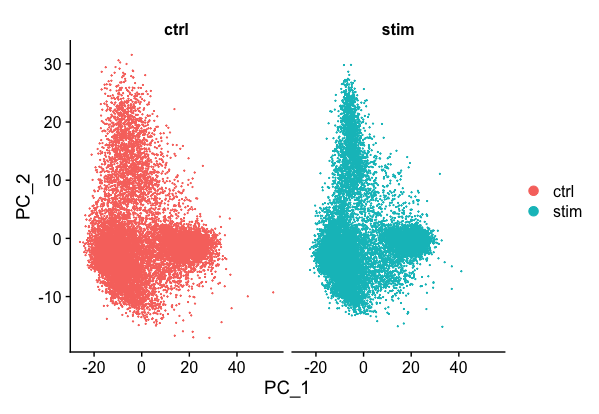
We can see with the PCA mapping that we have a good overlay of both conditions by PCA.
Now, we can also visualize with UMAP. Let’s run the method and plot. UMAP is a stochastic algorithm – this means that it makes use of randomness both to speed up approximation steps, and to aid in solving hard optimization problems. Due to the stochastic nature, different runs of UMAP can produce different results. We can set the seed to a specific (but random) number, and this avoids the creation of a slightly different UMAP each time re-run our code.
NOTE: Typically, the
set.seed()would be placed at the beginning of your script. In this way the selected random number would be applied to any function that uses pseudorandom numbers in its algorithm.
# Set seed
set.seed(123456)
# Run UMAP
seurat_integrated <- RunUMAP(seurat_integrated,
dims = 1:40,
reduction = "pca")
# Plot UMAP
DimPlot(seurat_integrated)
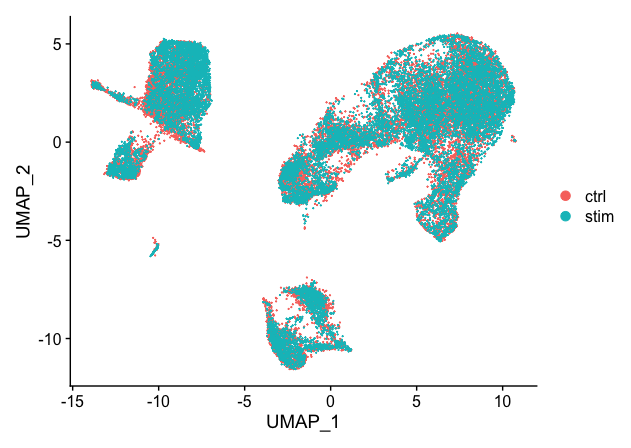
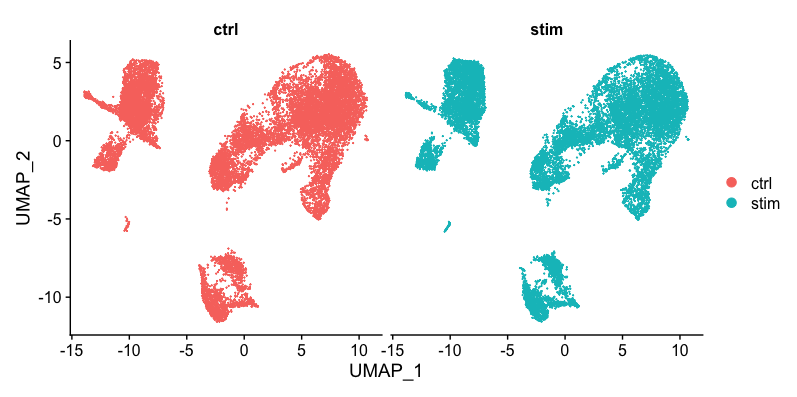
When we compare the similarity between the ctrl and stim clusters in the above plot with what we see using the the unintegrated dataset, it is clear that this dataset benefitted from the integration!
Side-by-side comparison of clusters
Sometimes it’s easier to see whether all of the cells align well if we split the plotting between conditions, which we can do by adding the split.by argument to the DimPlot() function:
# Plot UMAP split by sample
DimPlot(seurat_integrated,
split.by = "sample")
Save the “integrated” object!
Since it can take a while to integrate, it’s often a good idea to save the integrated seurat object.
# Save integrated seurat object
saveRDS(seurat_integrated, "results/integrated_seurat.rds")
Complex Integration Tasks
In the section above, we’ve presented the Seurat integration workflow, which uses canonical correlation analysis (CCA) and multiple nearest neighbors (MNN) to find “anchors” and integrate across samples, conditions, modalities, etc. While the Seurat integration approach is widely used and several benchmarking studies support its great performance in many cases, it is important to recognize that alternative integration algorithms exist and may work better for more complex integration tasks (see Luecken et al. (2022) for a comprehensive review).
Not all integration algorithms rely on the same methodology, and they do not always provide the same type of corrected output (embeddings, count matrix…). Their performance is also affected by preliminary data processing steps, including which normalization method was used and how highly variable genes (HVGs) were determined. All those considerations are important to keep in mind when selecting a data integration approach for your study.
What do we mean by a “complex” integration task?
In their benchmarking study, Luecken et al. (2022) compared the performance of different scRNA-seq integration tools when confronted to different “complex” tasks. The “complexity” of integrating a dataset may relate to the number of samples (perhaps generated using different protocols) but also to the biological question the study seeks to address (e.g. comparing cell types across tissues, species…). In these contexts, you may need to integrate across multiple confounding factors before you can start exploring the biology of your system.
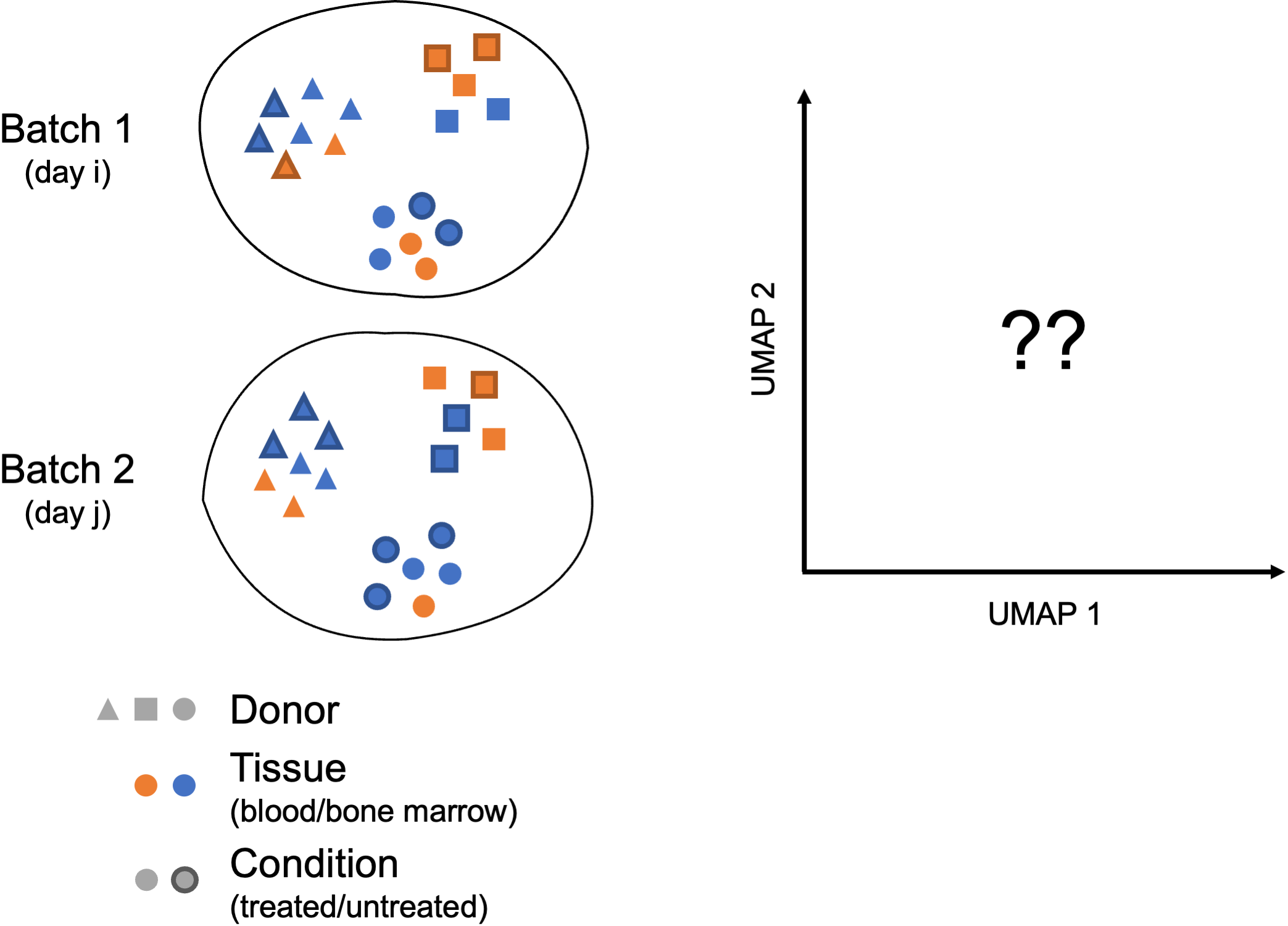
In these more complex scenarios, you want to select a data integration approach that successfully balances out the following challenges:
- Correcting for inter-sample variability due to source samples from different donors
- Correcting for variability across protocols/technologies (10X, SMART-Seq2, inDrop…; single-cell vs. single nucleus; variable number of input cells and sequencing depth; different sample preparation steps…)
- Identifying consistent cell types across different tissues (peripheral blood, bone marrow, lung…) and/or different locations (e.g. areas of the brain)
- Keeping apart cell subtypes (or even cell states) that show similar transcriptomes (CD4 naive vs. memory, NK vs NKT)
- Keeping apart cell subtypes that are unique to a tissue/condition
- Conserving the developmental trajectory, if applicable
Not all tools may perform as well on every task, and complex datasets may require testing several data integration approaches. You might want to analyze independently each of the batches you consider to integrate across, in order to define cell identities at this level before integrating and checking that the initially annotated cell types are mixed as expected.
Harmonizing as a method of integration
Harmony was devleoped in 2019, and is an example of a tool that can work with complex integration tasks. It is available as an R package on GitHub, and it has functions for standalone and Seurat pipeline analyses. It has been shown to perform incredibly well from recent benchmarking studies [1].
We have materials on Harmony and how to implement it within the Seurat workflow linked here, if you are interested in learning more.
This lesson has been developed by members of the teaching team at the Harvard Chan Bioinformatics Core (HBC). These are open access materials distributed under the terms of the Creative Commons Attribution license (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
- A portion of these materials and hands-on activities were adapted from the Satija Lab’s Seurat - Guided Clustering Tutorial