Learning Objectives
- Create new files and make edits to existing files with nano
- Compare different commands for examining small or large files
Creating text files
GUI text editors
You can easily create text files on your computer by opening up a text editor program such as TextWrangler, Sublime, and Notepad++, and start typing. These text editors often have features to easily search text, extract text, and highlight syntax from multiple programming languages. We refer to these as GUI text editors since they have a Graphical User Interface that has buttons and menus that you can click on to issue commands to the computer and you can move about the interface just by pointing and clicking.
NOTE: When we say, “text editor,” we really do mean “text”: these editors can only work with plain character data, not tables, images, or any other media and it explicitly excludes Microsoft Word or TextEdit.
Command-line text editors
But what if we need a text editor that functions from the command line interface? If we are working on remote computer (i.e. high-performance compute environments) we don’t have access to a GUI and so we need to use Command-line editors to create, modify and save files. When using these types of editors, you cannot ‘point-and-click’, you must navigate the interface using only the keyboard.
Some popular command-line editors include nano, Emacs or Vim. These editors are available by default on any shell environment, including on high-performance compute environments (local or cloud).
Introduction to Nano
Today, we are going to introduce the text editor ‘nano’. nano is simple to use and the most intuitive of the editors. It may not be as powerful in functionality, but it is a great start for those who are new to the command-line.
Nano Interface
You can create a document by calling a text editor and providing the name of the document you wish to create. Change directories to the unix_lesson/other folder and create a document using nano entitled draft.txt:
$ cd ~/unix_lesson/other
$ nano draft.txt
Now press enter.
Nano interface
You should see this:
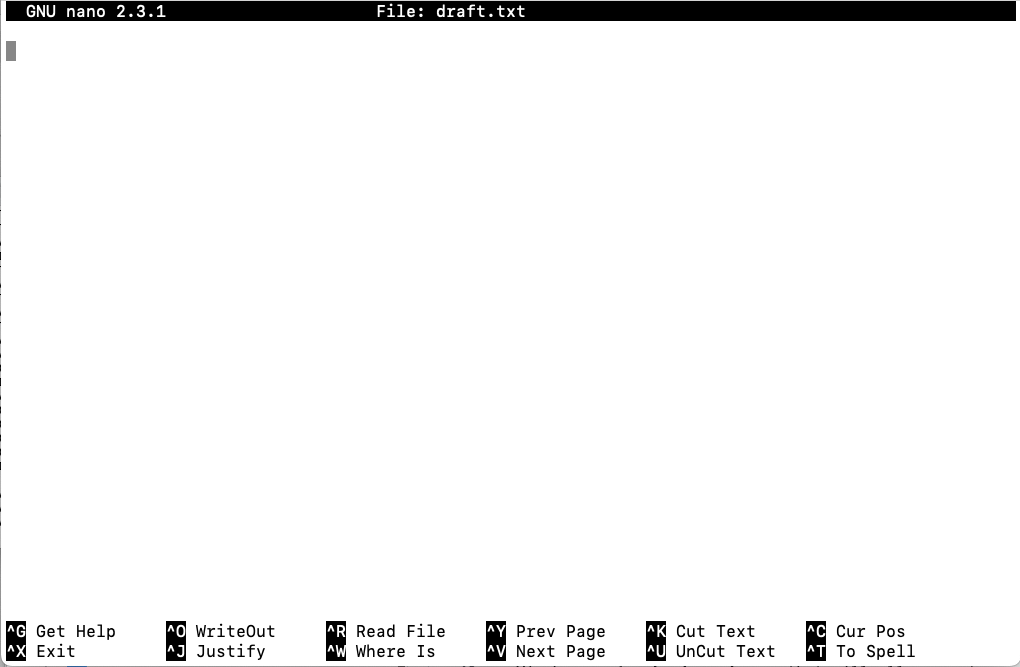
This should look quite similar to using a GUI plain text editor. We can see that our file is called draft.txt and our cursor is ready to add text! Type the following into the nanao editor window:
This is my first document.
This is a draft.
When I save this file it will be in my "other" folder.
The bottom of the screen provides a legend for the keystrokes that correspond to common commands that you may need when working in nano. To write to file (save), type Ctrl x. The program will ask if you are sure and you can type Y then press enter.
Open up the file again using the same command you used to create the file: nano draft.txt Do not modify the file close it again with Ctrl x. How is this different from the first time?
If you don’t change a file nano will not ask if you are sure to save it.
Nano Editing
Create the document spider.txt in nano. Copy and paste the text as follows:
The itsy bitsy spider
Went up the water spout
Down came the rain
And washed the spider out.
While we cannot point and click to navigate the document, we can use the arrow keys to move around. Navigating with arrow keys can be very slow, so nano has shortcuts. You can find some here. Several of the more common ones are already at the bottom of your nano window.
NOTE: If you are using a Mac you must change your terminal preferences in order to use the option button as an alt button. Go to: Terminal preferences -> Profiles -> (click keyboard button) -> select “Use Option as Meta key”
| Key | Action |
|---|---|
| Ctrl + k | Delete the current line |
| Ctrl + u | Paste |
Exercise
We have covered some basic commands in nano, but practice is key for getting comfortable with the program. Let’s practice what we just learned by folllowing the steps outlined below:
- Open
spider.txt, and delete line #2. - Quit without saving.
- Open
spider.txtagain, go to the last line and delete it. - Save the file and see whether your results below.
Answer
The itsy bitsy spider Went up the water spout Down came the rain
Examining Files
We now know how to move around the file system and look at the
contents of directories, and create files - but how do we look at the contents of files? Obviously we could open files up with a text editor like nano but there MUST be a better way!
The easiest way to examine a small file (i.e. few enough lines, that we could easily scroll through) is to just print out all of the contents using the command cat. Print the contents of spider.txt by entering the following command:
$ cat spider.txt
This prints out the all the contents of spider.txt to the screen.
catstands for catenate; it has many uses and printing the contents of a files onto the terminal is one of them.
spider.txt is all of 3 lines. When you have a 100000 line file you hardly want to print the entire thing to screen (and it can take a while!). When you are working on the cluster for data analysis you will inevitably encounter with large files.
For large files the command, less, can be useful. Let’s take a look at the files in the raw_fastq folder. You can check the size of them by using the command ls -lh. This will give you the size of them in human readable format.
$ cd ~/unix_lesson/raw_fastq
$ ls -lh
These files are quite large, so we probably do not want to use the cat command to look at them. Instead, we can use the less command. Let’s try this with one of the files.
less Mov10_oe_1.subset.fq
FASTQ files have four lines of data associated with every sequence read. Not only is there a header line and the nucleotide sequence, similar to a FASTA file, but FASTQ files also contain quality information for each nucleotide in the sequence.
The less command opens the file, and lets you navigate through it.
Shortcuts for less
| key | action |
|---|---|
| SPACE | to go forward |
| b | to go backwards |
| g | to go to the beginning |
| G | to go to the end |
| q | to quit |
less also gives you a way of searching through files. Just hit the / key to begin a search. Enter the name of the string of characters you would like to search for and hit enter. It will jump to the next location where that string is found. If you hit / then ENTER, less will just repeat the previous search. less searches from the current location and works its way forward. If you are at the end of the file and search for the word “cat”, less will not find it. You need to go to the beginning of the file and search.
For instance, let’s search for the sequence GAGACCC in our file. You can see that we go right to that sequence and can see what it looks like. To exit hit q.
There’s another way that we can look at files, and in this case, just look at part of them. This can be particularly useful if we just want to see the beginning or end of the file, or see how it’s formatted.
The commands are head and tail and they just let you look at
the beginning and end of a file respectively.
$ head Mov10_oe_1.subset.fq
$ tail Mov10_oe_1.subset.fq
Now that we have shown you how to create files, edit them and examin them - let’s move on to the next lesson to learn more!
This lesson has been developed by members of the teaching team at the Harvard Chan Bioinformatics Core (HBC). These are open access materials distributed under the terms of the Creative Commons Attribution license (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
- The materials used in this lesson were derived from work that is Copyright © Data Carpentry (http://datacarpentry.org/). All Data Carpentry instructional material is made available under the Creative Commons Attribution license (CC BY 4.0).
- Adapted from the lesson by Tracy Teal. Original contributors: Paul Wilson, Milad Fatenejad, Sasha Wood and Radhika Khetani for Software Carpentry (http://software-carpentry.org/)