Contributors: Meeta Mistry, Upendra Bhattarai, Heather Wick
Approximate time: 20 minutes
Learning Objectives
- Describe workflow management tools for bioinformatics analysis
- Overview of nf-core chipseq and understanding inputs and outputs
Workflow management for bioinformatics analysis
In the previous lesson we described the steps involved in going from raw sequence reads (FASTQ) to peak calls. Each step involves a different tool and typically is followed by some level of quality checks that follow. In this workshop, we provide lessons describing the workflow downstream of peak calls. It is important to thoroughly understand individual steps, the nuances of each tool, and the inputs and outputs. Therefore, when starting out on your analysis, running each command in the workflow is advantageous. However, as you start to scale up and you are running analyses on large datasets with many samples, using a workflow manager is something to consider.
Why use a workflow management system?
A workflow manager, in simple terms, is a tool or software that helps you organize and execute a series of tasks in a specific order. Below we outline some features of using a workflow manager:
- Modularity:
- The different steps in workflow are broken down into modular components (e.g., FASTQC, genome alignment)
- The workflow tasks are segregated from each other, each with its own specific inputs and outputs
- Modules can be reused in different analyses or modified to run the same analysis but tweaking certain parameters
- Automation:
- Once you have the complete workflow assembled, the process can easily be repeated on multiple samples and/or datasets
- Automating reduces the likelihood of human error and saves time
- Scalability:
- Workflow managers can efficiently handle large datasets and computationally intensive tasks, by distributing them across multiple processors or nodes
- Tasks are scheduled based on available resources, dependencies, and priorities
- Troubleshooting is more manageable because error-handling capabilities help in identifying failed processes and understanding the reasons behind failures
- Reproducibility:
- There is typically documentation, including metadata and provenenance tracking, providing clarity on what was done
- Transparency on software, parameter usage, and versions
Nextflow
Nextflow is an example of a commonly used workflow management system. It was originally developed at the Centre for Genomic Regulation in Spain and released as an open-source project on GitHub in March 2013. Since its inception, Nextflow has gained traction all over the world; it has a massive user community and is used by over 1,000 organizations, including some of the world’s largest pharmaceutical firms [1].
![]()
nf-core
In early 2017, a parallel community effort called nf-core was established. The nf-core project resulted in multiple groups and research institutes collaborating to develop and share high-quality, curated pipelines written in Nextflow. The nf-core ensures the reproducibility and portability of pipelines across different environments (e.g., local, HPC, Cloud), operating systems (e.g., Mac, Linux, Windows), and software versions.
For this workshop, the dataset was run through the nf-core/chip-seq workflow. In the metro map below you can see which modules are available to use at the different steps. We chose modules that align with our best practices (e.g., Bowtie2 for alignment, MACS2 for peak calling) and as such the some of the outputs from this pipeline are being used as inputs to the workshop.
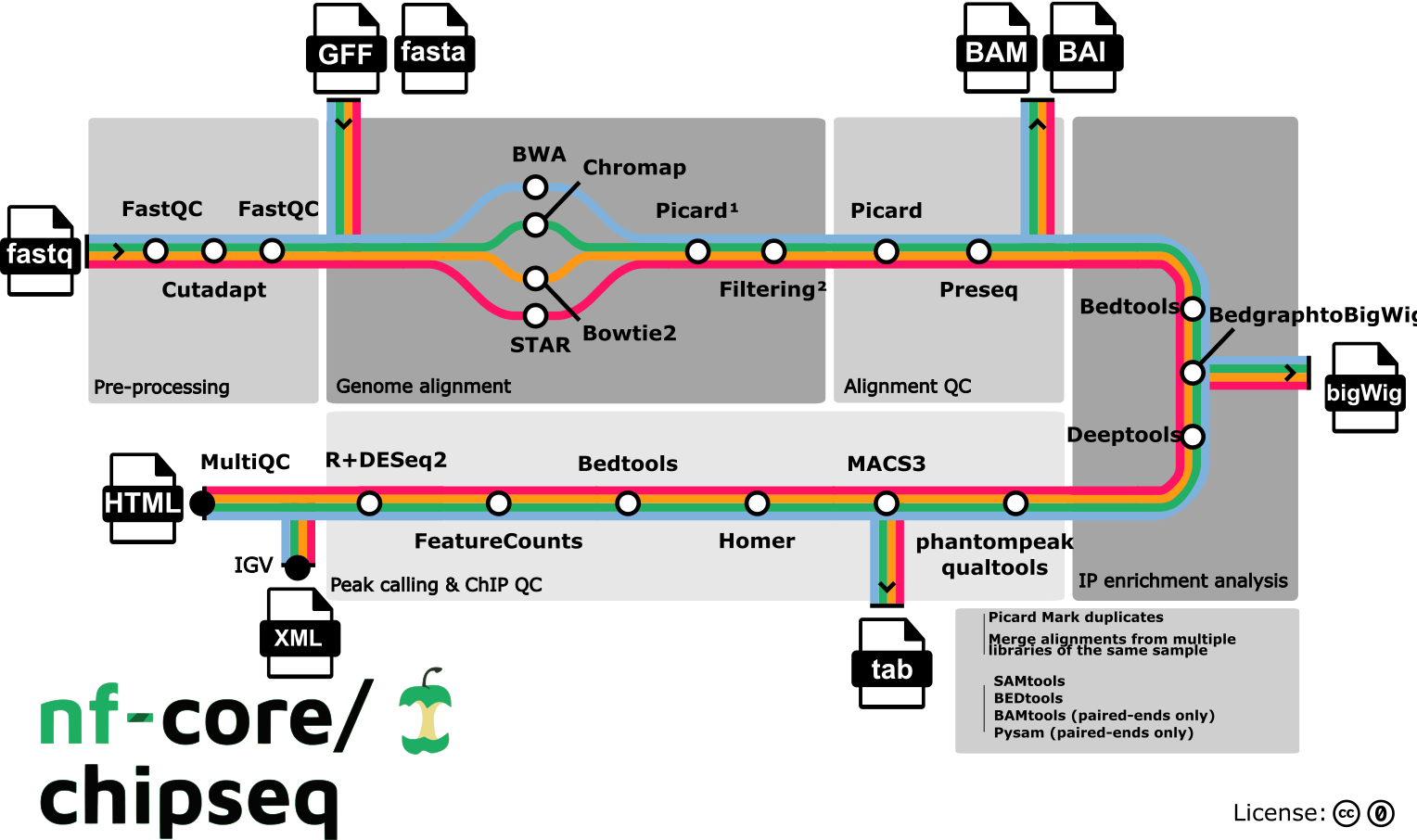
How can I learn to use nf-core?
Typically, scientific workflow systems initially present a steep learning challenge but once you figure out the basics things get easier. A good resource that we recommend is workshop created by the Carpentries, called “Introduction to Bioinformatics workflows with Nextflow and nf-core”. The Carpentries is a non-profit organization which teaches foundational coding and data science skills to researchers worldwide. This training can be done on your local computer or on a remote environment called Gitpod.
Resources
- Available nf-core pipelines
- nf-core tutorials
- An article on “The Story of Nextflow”
This lesson has been developed by members of the teaching team at the Harvard Chan Bioinformatics Core (HBC). These are open access materials distributed under the terms of the Creative Commons Attribution license (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.