Contributors: Meeta Mistry, Radhika Khetani
Approximate time: 75 minutes
Learning Objectives
- Combining replicates using simple overlap with Bedtools
Overlapping peaks
In this section, our goal is to determine what peaks are in common between the the two replicates for each factor (Nanog and Pou5f1). To perform this task we are going to use a suite of tools called bedtools.
bedtools
The idea is that genome coordinate information can be used to perform relatively simple arithmetic, like combining, subsetting, intersecting, etc., to obtain all sorts of information. bedtools from Aaron Quinlan’s group at University of Utah is easy to use, and an extremely versatile tool that performs tasks of this nature.

As the name implies, this suite of tools works with Bed files, but it also works with other file formats that have genome coordinate information.

NOTE: When working with multiple files to perform arithmetic on genomic coordinates, it is essential that all files have coordinate information for the same exact version of the genome and the same coordinate system (0-based or 1-based)!
Setting up
Let’s start an interactive session and change directories and set up a space for the resulting overlaps.
$ srun --pty -p short -t 0-12:00 --mem 8G --reservation=HBC bash
$ cd ~/chipseq/results/
$ mkdir bedtools
Load the modules for bedtools and samtools:
$ module load gcc/6.2.0 bedtools/2.26.0 samtools/1.3.1
Finding overlapping peaks between replicates
The bedtools intersect command within bedtools is the one we want to use, since it is able to report back the peaks that are overlapping with respect to a given file (the file designated as “a”).
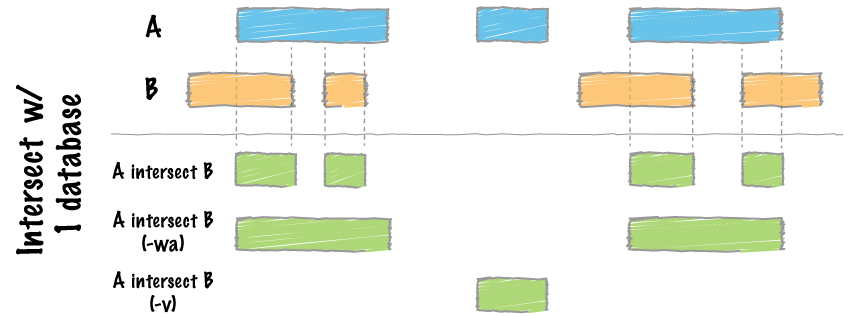
To find out more information on the parameters available when intersecting, use the help flag:
$ bedtools intersect -h
The intersect tool evaluates A (file 1) and finds regions that overlap in B (file 2). We will add the -wo which indicates to write the original A (file 1) and B (file 2) entries plus the number of base pairs of overlap between the two features.
Let’s start with the Nanog replicates:
$ bedtools intersect \
-a macs2/Nanog-rep1_peaks.narrowPeak \
-b macs2/Nanog-rep2_peaks.narrowPeak \
-wo > bedtools/Nanog-overlaps.bed
How many overlapping peaks did we get?
We’ll do the same for the Pou5f1 replicates:
$ bedtools intersect \
-a macs2/Pou5f1-rep1_peaks.narrowPeak \
-b macs2/Pou5f1-rep2_peaks.narrowPeak \
-wo > bedtools/Pou5f1-overlaps.bed
Note that we are working with subsetted data and so our list of peaks for each replicate is small. Thus, the overlapping peak set will be small as we found with both Nanog and Pou5f1. What is interesting though, is that even though the individual peak lists are smaller for Pou5f1 samples, the overlapping replicates represent a higher proportion of overlap with respect to each replicate.
Historical Note: “A simpler heuristic for establishing reproducibility was previously used as a standard for depositing ENCODE data and was in effect when much of the currently available data was submitted. According to this standard, either 80% of the top 40% of the peaks identified from one replicate using an acceptable scoring method should overlap the list of peaks from the other replicate, OR peak lists scored using all available reads from each replicate should share more than 75% of regions in common. As with the current standards, this was developed based on experience with accumulated ENCODE ChIP-seq data, albeit with a much smaller sample size.” [ENCODE Guidelines, Landt et al, 2012]
This lesson has been developed by members of the teaching team at the Harvard Chan Bioinformatics Core (HBC). These are open access materials distributed under the terms of the Creative Commons Attribution license (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.