Learning Objectives:
- Call variants with Freebayes
- Get familiar with the Variant Call Format (VCF)
- Use vcftools to perform some simple filtering on the variants in the VCF file
Variant Calling
We have the aligned and cleaned up the data, and have a BAM file ready for calling variants.
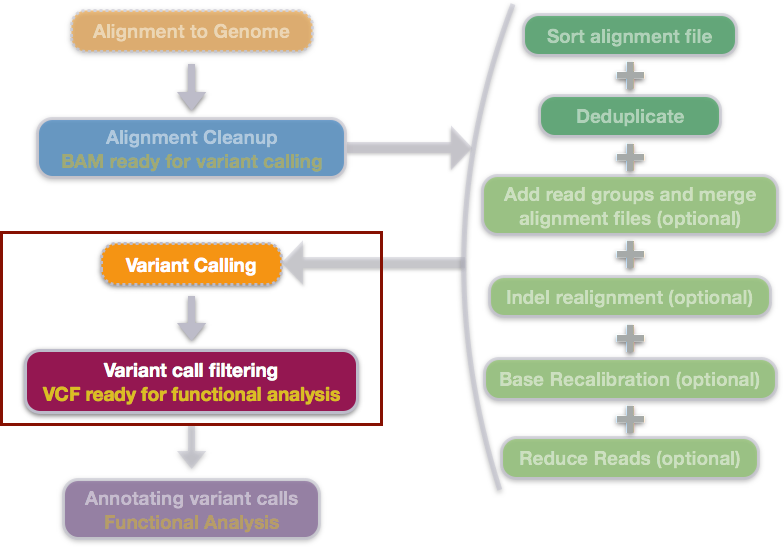
Some of the more popular tools for calling variants include SAMtools mpileup, the GATK suite and FreeBayes (Garrison and Marth, 2012). While it can be useful to work through the GATK Best Practices we will be using FreeBayes in this module as it is just as sensitive and precise, but has no license restrictions. After calling variants, we will filter out low quality variants using vcftools, a toolkit designed to work with Variant Call Format or VCF files.
Freebayes
FreeBayes is a haplotype-based variant detector and is a great tool for calling variants from a population.
“FreeBayes is a Bayesian genetic variant detector designed to find small polymorphisms, specifically SNPs (single-nucleotide polymorphisms), indels (insertions and deletions), MNPs (multi-nucleotide polymorphisms), and complex events (composite insertion and substitution events) smaller than the length of a short-read sequencing alignment.”
“FreeBayes is haplotype-based, in the sense that it calls variants based on the literal sequences of reads aligned to a particular target, not their precise alignment. This model is a straightforward generalization of previous ones (e.g. PolyBayes, samtools, GATK) which detect or report variants based on alignments. This method avoids one of the core problems with alignment-based variant detection— that identical sequences may have multiple possible alignments:”
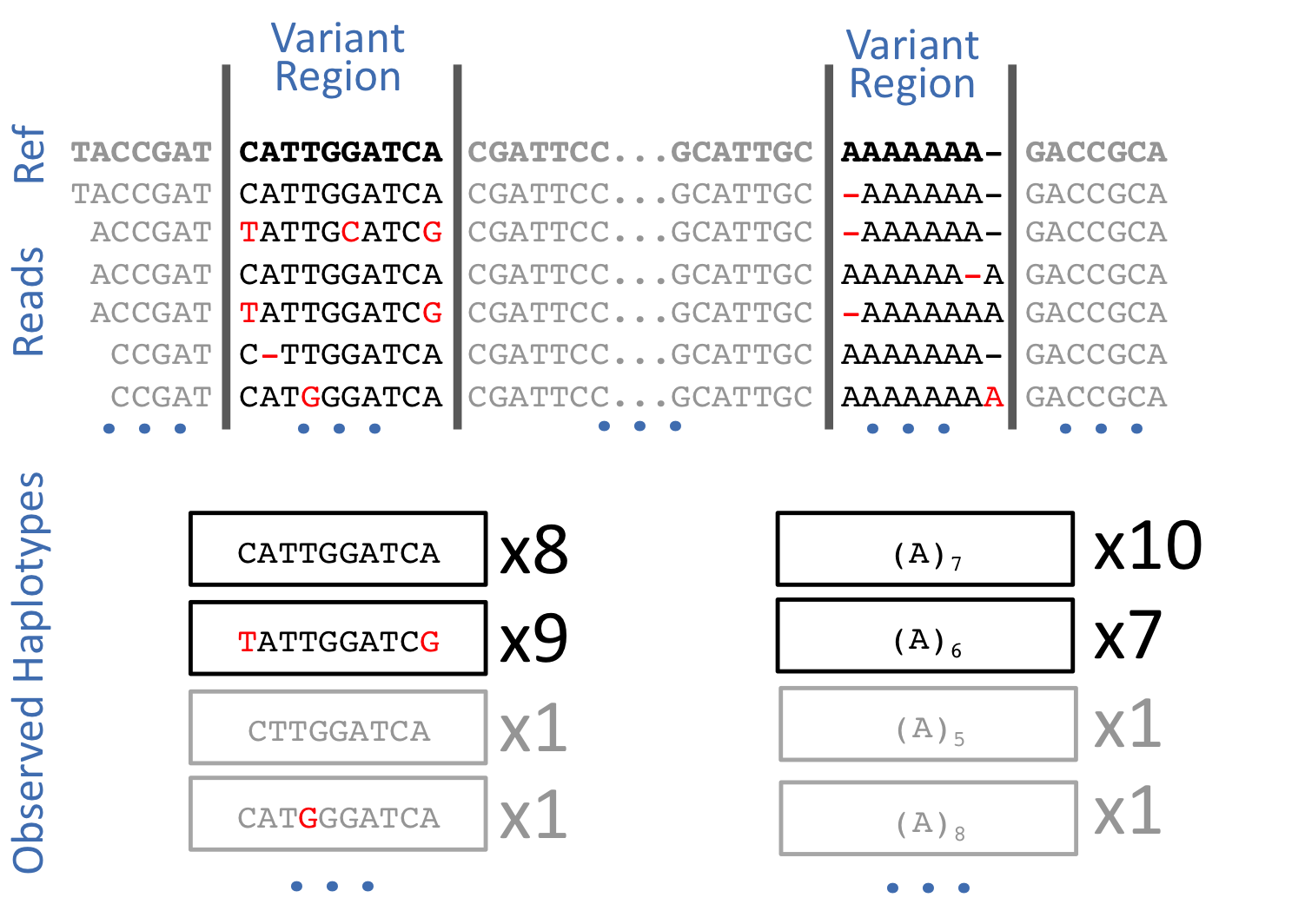

“FreeBayes uses short-read alignments (BAM files with Phred+33 encoded quality scores, now standard) for any number of individuals from a population and a reference genome (in FASTA format) to determine the most-likely combination of genotypes for the population at each position in the reference. It reports positions which it finds putatively polymorphic in variant call file (VCF) format. It can also use an input set of variants (VCF) as a source of prior information, and a copy number variant map (BED) to define non-uniform ploidy variation across the samples under analysis.”
Running FreeBayes
$ mkdir ~/var-calling/results/variants
$ cd ~/var-calling/results/variants/
$ which freebayes
If the output of the which command is /n/app/bcbio/tools/bin/freebayes, then you are all set! Since there is no module for freebayes on O2, we are using the bcbio version of the tool.
If you don’t get
/n/app/bcbio/tools/bin/freebayesas the output, do one of the following options below:Option #1:
$ PATH=:$PATHOption #2, add the following line to your
.bashrcfile:export PATH=/n/app/bcbio/tools/bin:$PATH
Once you have freebayes in your path, let’s check out our options:
$ freebayes -h
$ freebayes -f ~/var-calling/reference_data/chr20.fa ~/var-calling/results/bwa/na12878_sorted_marked.bam > ~/var-calling/results/variants/na12878.vcf
Variant Call Format (VCF)
VCF is a text format. It usually has several header lines before the actual data; the header lines start with ##. There is usually only 1 VCF file generated for all the samples in an experiment. Variants are represented in the rows, and each sample has a column with the status of a given variant:
##format=VCFv4.0
##fileDate=20090805
##source=myImputationProgramV3.1
##reference=1000GenomesPilot-NCBI36
##phasing=partial
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT NA00001 NA00002
20 14370 rs6054257 G A 29 0 NS=55;DP=255;AF=0.768;DB;H2 GT:GQ:DP:HQ 0|0:48:1:51,51 1|0:48:8:51,51
20 13330 . T A 3 q10 NS=55;DP=202;AF=0.024 GT:GQ:DP:HQ 0|0:49:3:58,50 0|1:3:5:65,3
20 1110696 rs6040355 A G,T 67 0 NS=55;DP=276;AF=0.421,0.579;AA=T;DB GT:GQ:DP:HQ 1|2:21:6:23,27 2|1:2:0:18,2
20 10237 . T . 47 0 NS=57;DP=257;AA=T GT:GQ:DP:HQ 0|0:54:7:56,60 0|0:48:4:51,51
20 123456 microsat1 G D4,IGA 50 0 NS=55;DP=250;AA=G GT:GQ:DP 0/1:35:4 0/2:17:2
Often the header lines will have some explanation about the various columns in the VCF, including the confusing looking INFO column. Here’s an explanation of the INFO column for the first entry in the example above (the example below is representing the same variant as above, “rs6054257”, but the VCF was excerpted from a much larger experiment):

Below is another example with slightly different fields in the INFO column:

Now, let’s take a look at the one we just generated:
$ less na12878.vcf
How does this compare to the 2 examples we have seen so far? How does the ID column compare?
Filtering VCFs
It is very important to filter out low-quality variants before moving to the assessment and annotation steps. Low quality variants usually represent sequencing errors (low quality bases). Freebayes variant quality determination is done as described here: https://github.com/ekg/freebayes#observation-qualities.
Today we are going to use vcftools to remove entries that have calls with a quality score of lower than 20.
$ module load gcc/6.2.0 vcftools/0.1.15
The manual for vcftools is available here, let’s take a quick look at it.
So the most basic options you need to specify are input --vcf <name> and output --out <name-filtered>. There are many different criteria that can be used for filtering the input vcf, below are a few examples.
Include/exclude specific sites by chromosome:
--chr 20
--not-chr 20
No two sites are within specified distance to one another:
--thin 5
Specify minimum depth for each site:
--minDP 10
Filter by variant type:
--keep-only-indels
--remove-indels
Include SNPs with specific ID (i.e. dbSNP, this is information we will be adding in the annotation section):
--snps <string>
We are going to stick with using only the quality score for today’s class:
$ vcftools --vcf na12878.vcf --minQ 20 --recode --recode-INFO-all --out na12878_q20
”
--recode: These options are used to generate a new file in either VCF or BCF from the input VCF or BCF file after applying the filtering options specified by the user. The output file has the suffix “.recode.vcf” or “.recode.bcf”. By default, the INFO fields are removed from the output file, as the INFO values may be invalidated by the recoding (e.g. the total depth may need to be recalculated if individuals are removed). This behavior may be overriden by the following options. By default, BCF files are written out as BGZF compressed files.””
--recode-INFO-all: These options can be used with the above recode options to define an INFO key name to keep in the output file. This option can be used multiple times to keep more of the INFO fields. The second option is used to keep all INFO values in the original file.”Information about
recodeadapted from the VCFtools manual.
Now we are (almost) ready to annotate this VCF with known information from dbSNP, and add functional annotation information to enable variant prioritization.
This lesson has been developed by members of the teaching team at the Harvard Chan Bioinformatics Core (HBC). These are open access materials distributed under the terms of the Creative Commons Attribution license (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.