Approximate time: 30 minutes
Learning Objectives
- Describe the best practices for designing a ChIP-seq experiment
- Recognize the need for data management and project organization
Introduction to ChIP-seq
Chromatin immunoprecipitation (ChIP) experiments are performed to identify DNA bound to specific (chromatin) proteins of interest. The first step involves isolating the chromatin and immunoprecipitating (IP) fragements with an antibody against the protein of interest. In ChIP-seq, the immunoprecipitated DNA fragments are then sequenced, followed by identification of enriched regions of DNA or peaks. These peak calls can then be used to make biological inferences by determining the associated genomic features and/or over-represented sequence motifs.
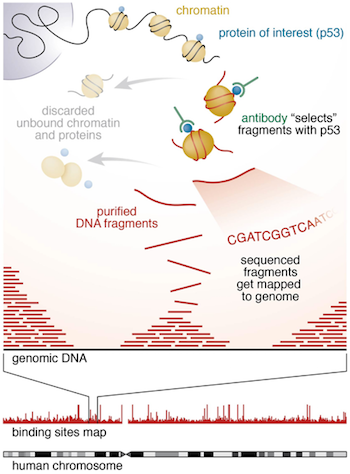
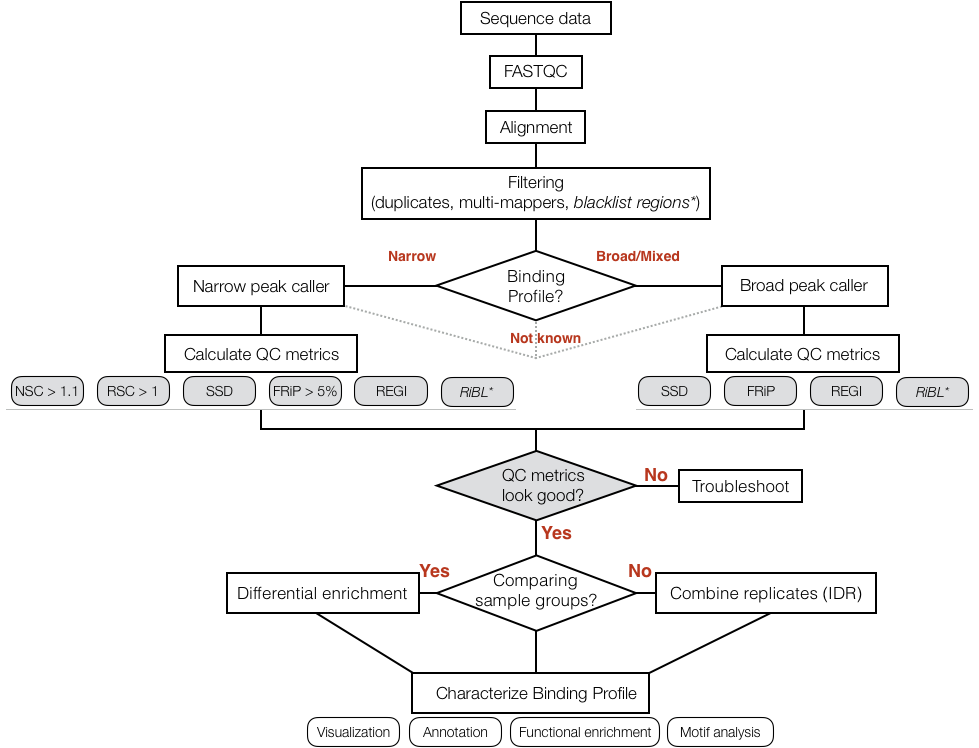
During this session we will be performing a complete workflow for ChIP-seq analysis, starting with the raw sequencing reads and ending with functional enrichment analyses and motif discovery.
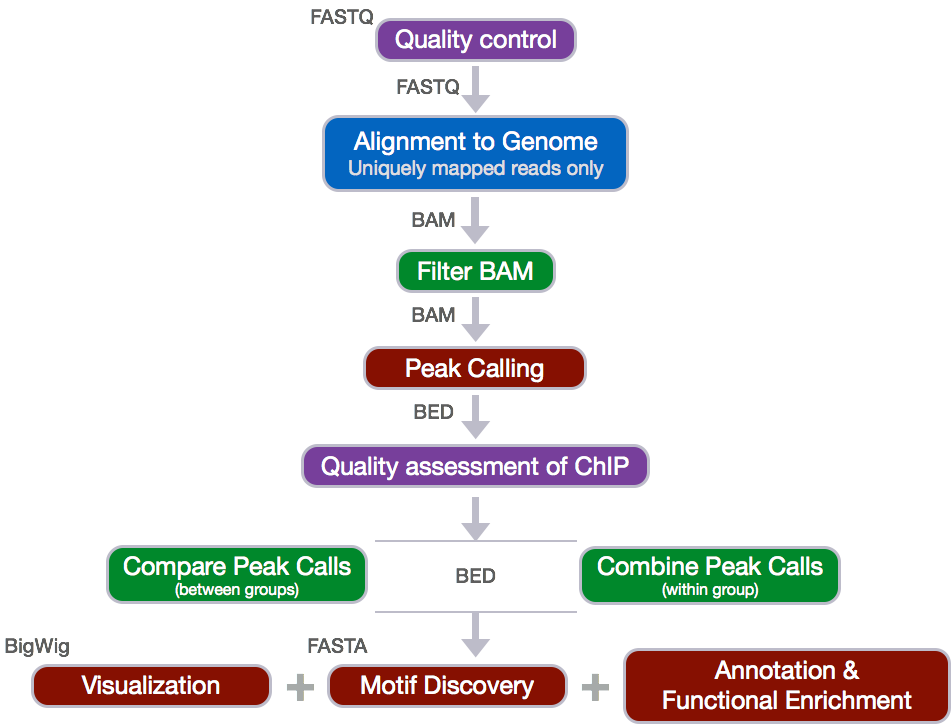
Decision Tree
For ChIP-seq the above workflow is a good guide to the analysis however, there are several steps along the way which require one to use different tools and assess metrics in a fairly subjective manner.
We will be going deeper into each component of the tree below as we introduce you to each step, but for now we wanted to give you a rough idea of what to expect as we go through the rest of the lessons for ChIP-seq.
Setting up
Since we are going to be working with this data on our remote server, O2, we first need to log onto the server.
Type in the following command with your username to login:
ssh username@o2.hms.harvard.edu
Next we will start an interactive session on O2 with 2 cores (add the -c 2):
$ srun --pty -p short -t 0-12:00 --mem 8G -c 2 --reservation=HBC bash
Let’s create a directory that we can use for the rest of the ChIP-seq session.
First, make sure that you are in your home directory.
$ cd
$ pwd
This should return /home/username.
Create a chipseq directory and change directories into it:
$ mkdir chipseq
$ cd chipseq
Now that we have a project directory, we can set up the following structure within it to keep files organized.
chipseq/
├── logs/
├── meta/
├── raw_data/
├── reference_data/
├── results/
│ ├── bowtie2/
│ └── fastqc/
└── scripts/
$ mkdir raw_data reference_data scripts logs meta
$ mkdir -p results/fastqc results/bowtie2
$ tree # this will show you the directory structure you just created
NOTE: that we are using the parents flag (
-por--parents) withmkdirto complete the file path by creating any parent directories that do not exist. In our case, we have not yet created theresultsdirectory and so since it does not exist it will be created. This flag can be very useful when scripting workflows.
This is a generic directory structure and can be tweaked based on personal preference and analysis workflow.
Now that we have the directory structure created, let’s copy over the data to perform our quality control and alignment, including our FASTQ files and reference data files:
$ cp /n/groups/hbctraining/chip-seq/raw_fastq/*fastq raw_data/
$ cp /n/groups/hbctraining/chip-seq/reference_data/chr12* reference_data/
Now we are all set up for our analysis!
Exploring the dataset
Our goal for this session is to compare the the binding profiles of Nanog and Pou5f1 (Oct4). The ChIP was performed on H1 human embryonic stem cell line (h1-ESC) cells, and sequenced using Illumina. The datasets were obtained from the HAIB TFBS ENCODE collection. For these 6 samples, we will be using reads from only a 32.8 Mb of chromosome 12 (chr12:1,000,000-33,800,000), so we can get through the workflow in a reasonable amount of time.
Two replicates were collected and each was divided into 3 aliquots for the following:
- Nanog IP
- Pou5f1 IP
- Control input DNA
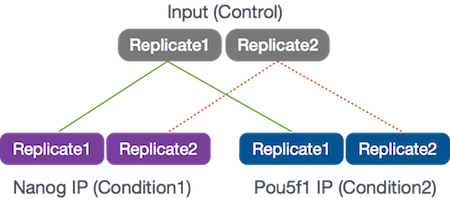
These 2 transcription factors are involved in stem cell pluripotency and one of the goals is to understand their roles, individually and together, in transriptional regulation.
This lesson has been developed by members of the teaching team at the Harvard Chan Bioinformatics Core (HBC). These are open access materials distributed under the terms of the Creative Commons Attribution license (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.