Accessing genomic reference data
During an NGS experiment, the nucleotide sequences stored inside the raw FASTQ files, or “sequence reads”, need to be mapped or aligned to the reference genome to determine from where these sequences originated. Therefore, we need a reference genome (in FASTA format) in which to align our sequences.
In addition, many NGS methods require knowing where known genes or exons are located on the genome in order to quantify the number of reads aligning to different genome features, such as exons, introns, transcription start sites, etc. These analyses require reference data containing specific information about genomic coordinates of various genomic “features”, such as gene annotation files (in GTF, GFF, etc.).
To download reference data, there are a few different sources available:
- General biological databases: Ensembl, NCBI, and UCSC
- Organism-specific biological databases: Wormbase, Flybase, etc. (often updated more frequently, so may be more comprehensive)
- Reference data collections: Illumina’s iGenomes, one location to access genome reference data from Ensembl, UCSC and NCBI
- Local access: shared databases on the FAS-RC cluster or HMS-RC’s O2 cluster with access to genome reference data from Ensembl, UCSC and NCBI
*Note that these reference data sources are relevant to most types of genomic analyses, not just NGS analyses.
General biological databases
Biological databases for gene expression data store genome assemblies and provide annotations regarding where the genes, transcripts, and other genomic features are located on the genome.
Genome assemblies give us the nucleotide sequence of the reference genome. Although the Human Genome Project was “completed” in 2003, small gaps in the sequence remained (estimated 1% of gene-containing portions). As technology improves and more genomes are sequenced, these gaps are filled, mistakes are corrected and alternate alleles are provided. Therefore, every several years a new genome build is released that contains these improvements.
The current genome build is GRCh38/hg38 for the human, which was released in 2013 and is maintained by the Genome Reference Consortium (GRC). Usually the biological databases will include the updated versions as soon as they are stably released, in addition to access to archived versions.
Genome databases incorporate these genomes and generate the gene annotations with the following similarities/differences:
- Ensembl, NCBI, and UCSC all use the same genome assemblies or builds provided by the GRC
- GRCh38 = hg38; GRCh37 = hg19
- Patches or minor revisions of the genome, which don’t change the genome coordinates, are frequently provided by the GRC. Each database makes the patches available for users at different intervals. If the user applies the patches, the genome reference sequence may differ between databases.
- GRCh38p1 != GRCh38p2
-
Each biological database independently determines the gene annotations; therefore, gene annotations between these databases can differ, even though the genome assembly is the same. Naming conventions are also different (chr1=1) between databases.
- Always use the same biological database for all reference data!
Ensembl
Ensembl provides a website that acts as a single point of access to annotated genomes for vertebrate species. For all other organisms there are additional Ensembl databases available through Ensembl Genomes; however, they do not include viruses (NCBI does).
- Genome assemblies/builds (reference genomes)
- New genome builds are released every several years or more depending on the species
- Genome assemblies are updated every two years to include patches, or less often depending on the species
- Gene annotations
- Gene annotations are created or updated using a variety of sources (ENA, UniProtKB, NCBI RefSeq, RFAM, miRBase, and tRNAscan-SE databases)
- Automatic annotation is performed for all species using identified proteins and transcripts
- Manual curation by the HAVANA group is performed for human, mouse, zebrafish, and rat species, providing better confidence of transcript annotations
- Directly imports annotations from FlyBase, WormBase and SGD
Using the Ensembl genomic database and genome browser
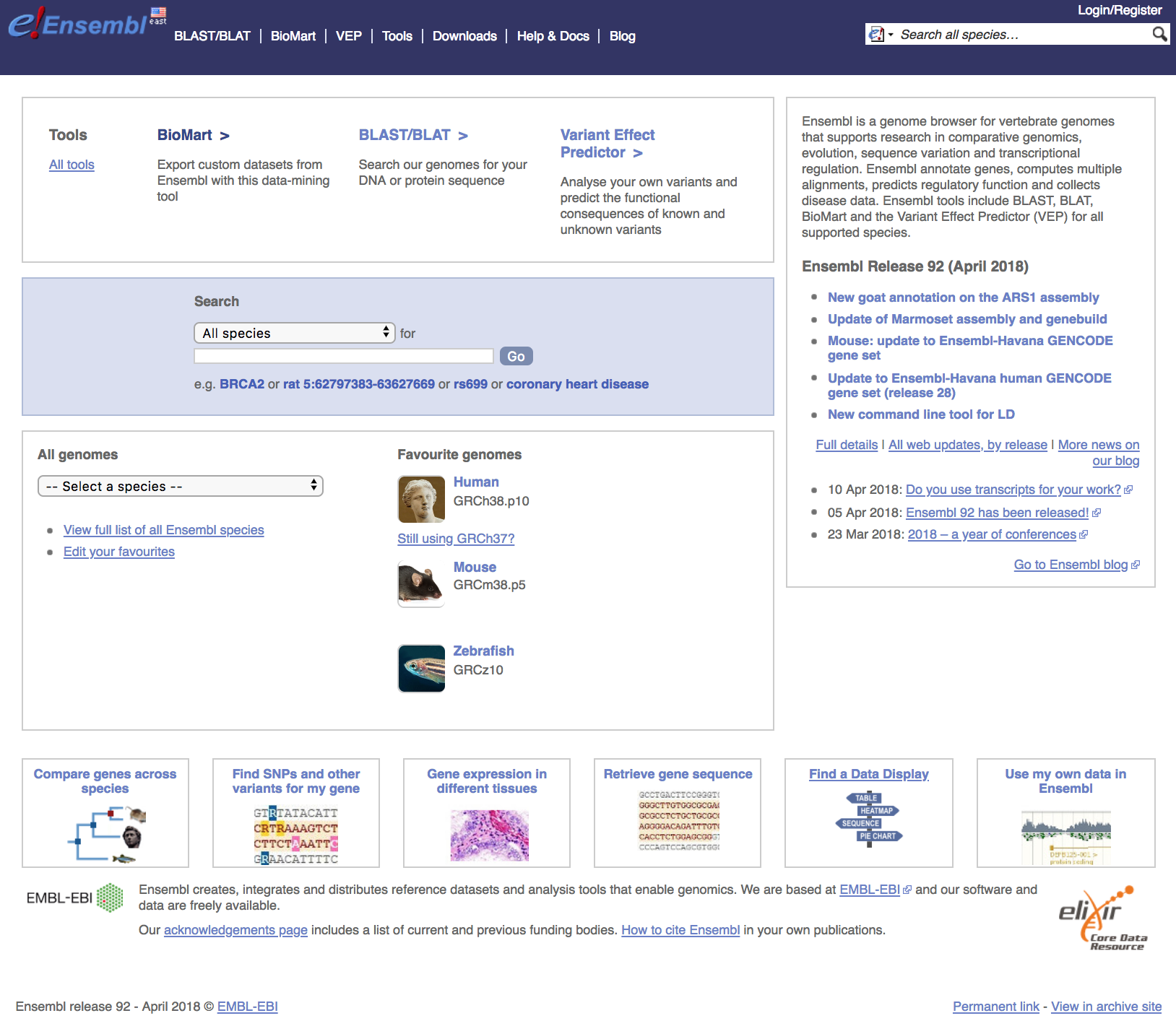
Navigate to the Ensembl website to view the interface. The homepage for Ensembl has a lot to offer, with the a lot of information and access to a range of functionality and tools.
-
Searching Ensembl: Look for a gene, location, variant and more using the search box on the homepage or the box that is provided in the top right corner of any Ensembl page.
- a gene name (for example, BRCA2) - best to use the official gene symbols (HGNC)
- a UniProt accession number (for example, P51587)
- a disease name (for example, coronary heart disease)
- a variation (for example, rs1223)
- a location - a genomic region (for example, rat X:100000..200000)
- a PDB ID or a Gene Ontology (GO) term
Most search results will take you to the appropriate Ensembl view through a results page. These linked pages will allow you to download information/sequences for specific genes/transcripts/exons/variants. If you search using a location you will be directed straight to the location tab (this tab provides a view of a region of a genome).
-
Browse a Genome: Choose your species of interest in this section. The drop down menu under ‘All genomes’ allows you to select from the full list. The Ensembl Pre! site contains new genomes (either new species to Ensembl or updates in the reference assembly) that do not yet have an Ensembl gene set. BLAST/BLAT is available for organisms in all Ensembl sites, including Pre!
-
Help: There is a wealth of help and documentation in Ensembl if you are new to the browser. Video tutorials are provided and printable pdfs with exercises. Custom data may be uploaded to Ensembl or displayed directly by attaching a file by URL.
-
News: To find out what genome build and release you are working with, have a look at the news section of the homepage. If the current release is not the one you need, access archive sites to access previous versions, or releases, of Ensembl using the link on the lower right side.
While we are not going to explore the Ensembl database in this workshop, we have materials available if you wish to explore on your own.
Ensembl identifiers
When using Ensembl, note that it uses the following format for biological identifiers:
- ENSG###########: Ensembl Gene ID
- ENST###########: Ensembl Transcript ID
- ENSP###########: Ensembl Peptide ID
- ENSE###########: Ensembl Exon ID
For non-human species a suffix is added:
- ENSMUSG###: MUS (Mus musculus) for mouse
- ENSDARG###: DAR (Danio rerio) for zebrafish
Finding and accessing reference data on Ensembl
The interface for downloading reference data from Ensembl is straight-forward. On the home page, you can click on Downloads.

Then click on the section to Download a sequence or region.

In the ‘Export Data’ window, click on the link for the FTP site.
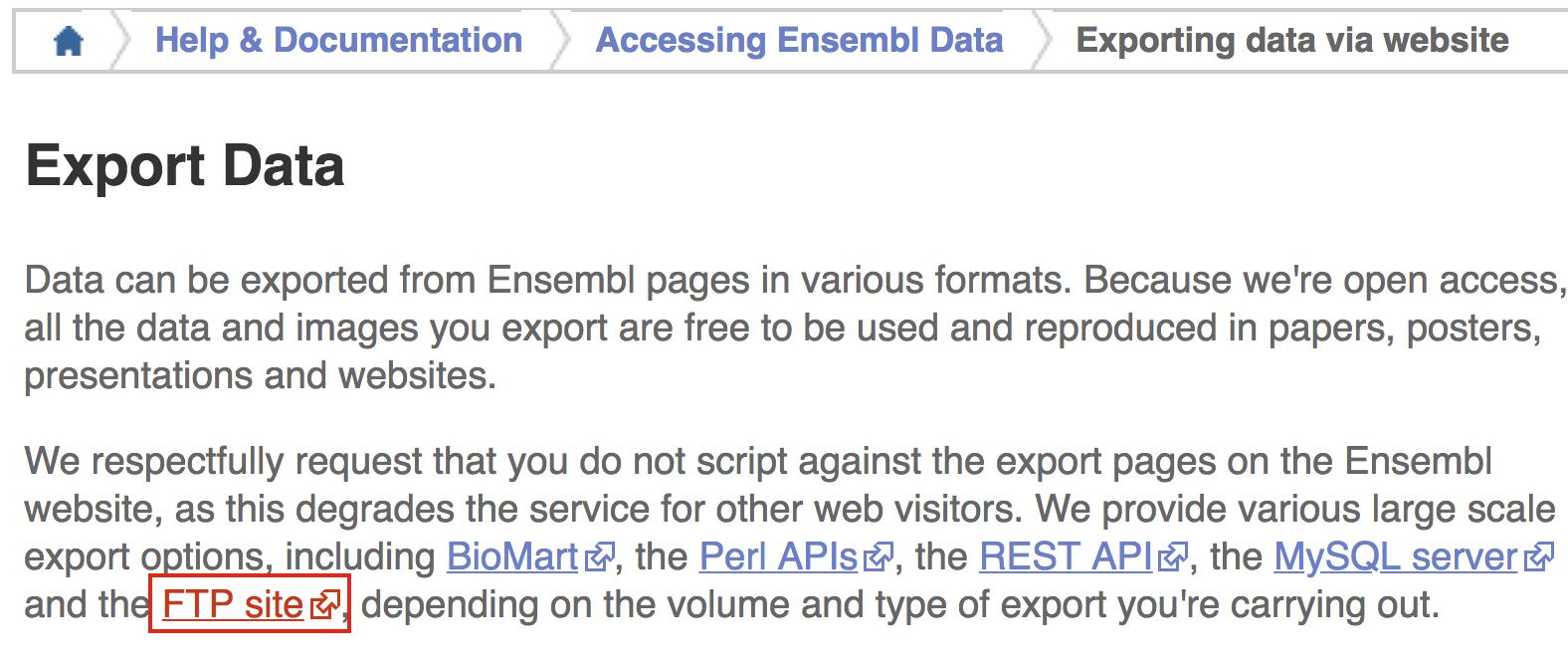
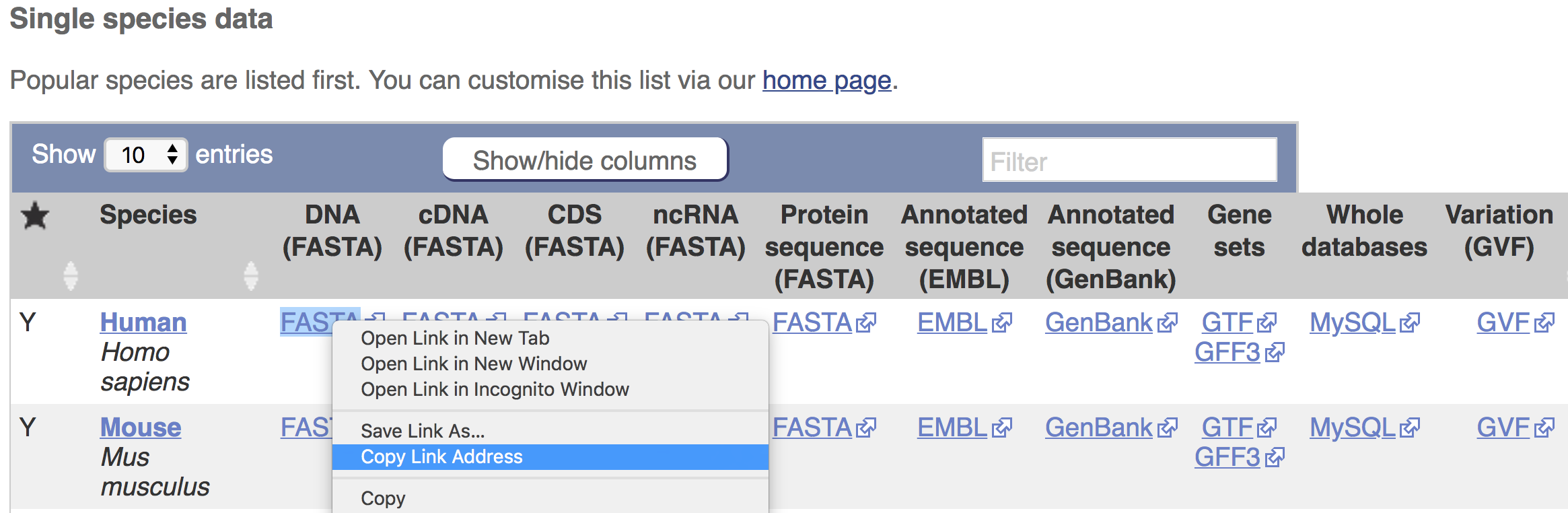
Finally, right-click on the link to the reference genome (DNA FASTA), reference transcriptome (cDNA FASTA), gene annotation file (Gene sets, GTF or GFF), or other required reference data to download. Copy the link address.
Now, on an HPC environment (O2 or FAS-RC cluster) you could use the wget command to download the reference data:
## DO NOT RUN
$ wget ftp://ftp.ensembl.org/pub/release-92/fasta/homo_sapiens/dna/
This would take a really long time for large genomes, so instead, you would probably want to submit a batch job using a script similar to the one below:
#!/bin/bash
#SBATCH -p short # partition name (small partition on O2)
#SBATCH -t 0-6:00 # hours:minutes runlimit after which job will be killed
#SBATCH -c 1 # number of cores requested
#SBATCH -o %J.out # File to which standard out will be written
#SBATCH -e %J.err # File to which standard err will be written
wget ftp://ftp.ensembl.org/pub/release-92/fasta/homo_sapiens/dna/
NOTE: If you desired an archived version of the genome, on the Ensembl home page for the organism of interest you would click on the
View in archive sitelink in the lower right-hand corner of the page. Then you would navigate as described above.
To run the script, you would use the following sbatch command:
## DO NOT RUN
$ sbatch name_of_script.slurm
Genomic reference data could be downloaded similarly by FTP from the NCBI FTP (or through Aspera) or the UCSC FTP.
Reference data collections: iGenomes
If working on a commonly analyzed organism, Illumina’s iGenomes has facilitated the process of downloading reference data. On the iGenomes website reference data from Ensembl, UCSC and NCBI for various genome builds are available for download. In addition, the download is a compressed file containing the matching reference genome (FASTA) and gene annotation (GTF/GFF) files.
To download from iGenomes, we can right-click and copy the link to the file and use our wget command.
#!/bin/bash
#SBATCH -p shared # partition name (small partition on O2)
#SBATCH -t 0-6:00 # hours:minutes runlimit after which job will be killed
#SBATCH -n 1 # number of cores requested
#SBATCH -o %J.out # File to which standard out will be written
#SBATCH -e %J.err # File to which standard err will be written
$ wget ftp://igenome:G3nom3s4u@ussd-ftp.illumina.com/Homo_sapiens/NCBI/GRCh38/Homo_sapiens_NCBI_GRCh38.tar.gz
After running the script with the sbatch command, you could use the tar command to unpack it.
$ tar -xzf Homo_sapiens_NCBI_GRCh38.tar.gz
If desired you could just run the tar command after the wget command in the above script to automatically unpack the reference data.
Local access via FAS-RC cluster or O2
Downloading the reference data from biological database or iGenomes might not be necessary since the Harvard FAS-RC cluster and O2 clusters have shared reference data downloaded from iGenomes available to its users.
On O2 shared data is located at /n/groups/shared_databases/igenome/. Instead of using storage space inside your folder, give the path to the reference data in these shared databases instead.
Let’s explore what’s available within the igenome folder and how to find the reference sequence and gene annotation files.
$ cd /n/groups/shared_databases/igenome/
Organism-specific databases
Although the general genomic databases update the genome builds and annotations for all species, organism-specific databases often update the genome patches and gene annotations more frequently. In addition, these databases also offer genomes for other species that may not be present in the general databases. Additional tools and information regarding these organisms are also accessible.
Wormbase
As an example, we will explore WormBase ParaSite, which is devoted to the study of C. elegans and other nematodes, in addition to, helminths. This site is closely linked with WormBase, and incorporates the information for C. elegans and other nematodes from this repository.
On the homepage, there is direct access to WormBase, links to all species genomes available, tools, and news.
Finding and accessing reference data on Wormbase
Downloading reference data from WormBase ParaSite is intuitive and simple. All that is needed is to click on the Downloads tab.

This will take you to the FTP site, where you can right-click to copy the link address of the reference data of interest.
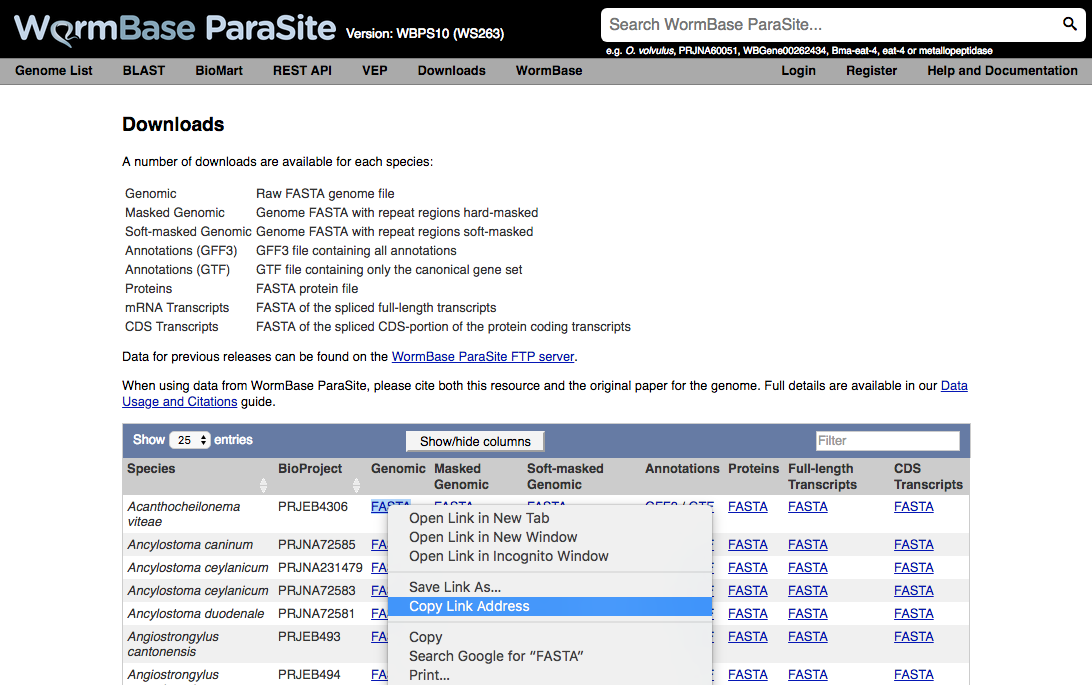
Then, similar to the other methods, the wget command can be used to download to the cluster.
#!/bin/bash
#SBATCH -p short # partition name (small partition on O2)
#SBATCH -t 0-6:00 # hours:minutes runlimit after which job will be killed
#SBATCH -c 1 # number of cores requested
#SBATCH -o %J.out # File to which standard out will be written
#SBATCH -e %J.err # File to which standard err will be written
wget ftp://ftp.ebi.ac.uk/pub/databases/wormbase/parasite/releases/WBPS10/species/acanthocheilonema_viteae/PRJEB4306/acanthocheilonema_viteae.PRJEB4306.WBPS10.genomic.fa.gz
In general, whether downloading data from a general database, an organism-specific database, or an iGenomes collection, the download should be straight-forward using the wget command and the link to the corresponding FTP site.
$ wget link_to_FTP
This lesson has been developed by members of the teaching team at the Harvard Chan Bioinformatics Core (HBC). These are open access materials distributed under the terms of the Creative Commons Attribution license (CC BY 4.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.